Understanding ion transport mechanisms in plants is crucial for enhancing stress tolerance and crop yields. The Arabidopsis thaliana High-Affinity Potassium Transporter 1; 1 (AtHKT1; 1) is vital for maintaining sodium and potassium balance under saline stress. However, the structural and functional effects of genetic variants in AtHKT1; 1, especially in potassium chloride (KCl) environments, are not fully understood. This research combiness Genome-Wide Association Studies (GWAS) with CRISPR-based functional validation to examine AtHKT1; 1 gene variants, focusing on the protein structure with PDB ID: 8W9O. The study first used GWAS and SNP discovery to find significant genetic variations linked to ion transport and salt tolerance. Candidate SNPs were selected based on their statistical significance and potential biological roles. Structural analysis of the protein 8W9O involved PDB and MMDB resources, with validation through ERRAT and visualization/mutation mapping in PyMOL. InterProScan was used to identify conserved functional motifs. To validate SNP effects, CRISPR guide RNAs were designed with E-CRISP and CHOPCHOP, targeting key gene regions for precise editing. This integrated approach linked genetic variations to structural changes and their potential impact on ion binding and transport, especially under KCl conditions. Results showed certain SNPs cause conformational shifts in key transmembrane regions of AtHKT1; 1, possibly influencing ion selectivity and transport efficiency. Structural validation confirmed the accuracy of the modeled variants, and domain analysis revealed disruptions in conserved functional areas. CRISPR strategies proved feasible for precise gene editing to test these functional hypotheses. Overall, this study offers a comprehensive framework that combines GWAS, structural bioinformatics, and CRISPR technology to explore how genetic variants affect AtHKT1; 1 function. These insights improve understanding of ion transport regulation in saline environments and support the development of genetically modified crops with enhanced salt tolerance.
| Published in | Computational Biology and Bioinformatics (Volume 14, Issue 1) |
| DOI | 10.11648/j.cbb.20261401.13 |
| Page(s) | 26-40 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Arabidopsis Thaliana, GWAS, SNPs, CRISPR-Cas9, Ion Transport, KCl Stress, Salt Tolerance Mechanism
Molecule name | Sodium Transporter HKT1 |
|---|---|
Classification | Transport Protein |
Secondary Structure | PDB Data Records |
Database Code | 8W9O |
Experiment Technique | Electron Microscopy |
Number of Chains | 4 |
Number of Groups | 854 (4) |
Number of Total Atoms | 6862 (4) |
Number of Carbon atoms | 4570 |
Number of Oxygen atoms | 1190 |
Number of Nitrogen atoms | 1058 |
Number of Hydrophobic atoms | 3780 |
Number of Helix atoms | 4964 |
Number of Sheet atoms | 82 |
Number of Backbone atoms | 3416 |
Number of Sidechain atoms | 3442 |
Number of Bonds | 7032 |
Number of Helices | 43 |
Number of Strands | 4 |
Number of Turns | 0 |
Number of H-Bonds | 588 |
Number of water molecule | 0 |
Protein type | Hydrophobic Protein (non-polar - 3572 atoms) |
Maximum Amino Acid Residue | Leucine (976 atoms) |
Minimum Amino Acid Residue | Cysteine (96 atoms) |
GO Category | GO Term | Description |
|---|---|---|
Molecular Function (MF) | GO: 0015079 | Potassium ion transmembrane transporter activity |
Biological Process (BP) | GO: 0006813 | Potassium ion transport |
Cellular Component (CC) | GO: 0005886 | Plasma membrane |
| [1] | Baxter I, Brazelton JN, Yu D, et al. “A coastal cline in sodium accumulation in Arabidopsis thaliana is driven by natural variation of the sodium transporter AtHKT1; 1.” PLoS Genetics. 2010; 6(11): e1001193. |
| [2] | D. An, et al. “AtHKT1 drives adaptation of Arabidopsis thaliana to salinity.” Nature Communications. 2017. |
| [3] | Huang, L., Wu, D. Z., & Zhang, G. P. (2020). Advances in studies on ion transporters involved in salt tolerance and breeding crop cultivars with high salt tolerance. Journal of Zhejiang University-SCIENCE B, 21(6), 426-441. |
| [4] | Angon, P. B., Mondal, S., Akter, S., Sakil, M. A., & Jalil, M. A. (2023). Roles of CRISPR to mitigate drought and salinity stresses on plants. Plant Stress, 8, 100169. |
| [5] | Dr Uma kumari, Devanshi Gupta, In silico RNA aptamer drug design and modelling, 2022/4, Journal-JETIR, Volume-9, Issue-4, Pages 718-725 Dr Uma kumari, Devanshi Gupta, In silico RNA aptamer drug design and modelling, 2022/4, Journal-JETIR, Volume-9, Issue-4, Pages 718-725 |
| [6] | Kumari, Uma &Tanwar, Aastha& George, Jositta&Nayak, Daityari. (2023). NGS Analysis to Detect Mutation in Brain Tumor Diagnostic. International Journal for Research in Applied Science and Engineering Technology. 11. |
| [7] | Craig PA, Michel LV, Bateman RC. A survey of educational uses of molecular visualization freeware. BiochemMolBiol Educ. 2013 May-Jun; 41(3): 193-205. |
| [8] | Johri, V., Bandbe, T., Kumari, U. (2026). CRISPR-CAS 9 Modeling of ALK Resistance Mutations Harbouring the G1202R/L1196M. American Journal of BioScience, 14(1), 8-19. |
| [9] | Bandbe, T., Johri, V., Kumari, U. (2025). Structure-guided Genome-wide Association Analysis of ALK Variants with GWAS Data Using R. Computational Biology and Bioinformatics, 13(2), 72-85. |
| [10] | Uma Kumari Rechel Tirkey Vipasha Rathi “Engineering Probiotic Strains for Gut Health Enhancement Using CRISPR and Molecular Marker-assisted Technologies” Published in Computational Biology and Bioinformatics (Volume 14, Issue 1) Received: 18 December 2025 Accepted: 29 December 2025 Published: 19 January 2026; |
| [11] | Uma Kumari, Sumita Katal, Shivangi Koundal; Structure-guided CRISPR CAS9 Targeting of ABL1 for Functional Disruption of BCR-ABL1 Fusion in Chronic Myeloid Leukaemia Publication, Computational Biology and Bioinformatics, Volume 14, Issue 1, 2026 Received: 10 January 2026 February, Accepted: 21 January 2026 Published: 4 February 2026. |
| [12] | Wang, X., Zhang, Z. X., Wang, W. X., Li, S. T., & Wang, Y. X. (2024). Functional identification of CCR1 gene enhancing saline-alkali stress tolerance. BMC Plant Biology, 24, 215. |
| [13] | Fussy, A., & Papenbrock, J. (2024). Molecular responses of Salicornia europaea to salinity stress. International Journal of Molecular Sciences, 25(9), 5021. |
| [14] | Popova, L. G., Khramov, D. E., Nedelyaeva, O. I., & Volkov, V. S. (2023). Yeast heterologous systems for studying plant membrane transport proteins. International Journal of Molecular Sciences, 24(12), 9987. |
| [15] | Sher, A., Nawaz, A., Ul-Allah, S., Sattar, A., & Manaf, A. (2024). Role of 5-aminolevulinic acid in improving salt tolerance in sunflower. Acta Physiologiae Plantarum, 46(2), 89. |
APA Style
Sachan, S., Kumari, U. (2026). Integrative GWAS and CRISPR Functional Validation of Gene Variant Structure of AtHKT1; 1 in KCl. Computational Biology and Bioinformatics, 14(1), 26-40. https://doi.org/10.11648/j.cbb.20261401.13
ACS Style
Sachan, S.; Kumari, U. Integrative GWAS and CRISPR Functional Validation of Gene Variant Structure of AtHKT1; 1 in KCl. Comput. Biol. Bioinform. 2026, 14(1), 26-40. doi: 10.11648/j.cbb.20261401.13
@article{10.11648/j.cbb.20261401.13,
author = {Shephali Sachan and Uma Kumari},
title = {Integrative GWAS and CRISPR Functional Validation of Gene Variant Structure of AtHKT1; 1 in KCl},
journal = {Computational Biology and Bioinformatics},
volume = {14},
number = {1},
pages = {26-40},
doi = {10.11648/j.cbb.20261401.13},
url = {https://doi.org/10.11648/j.cbb.20261401.13},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cbb.20261401.13},
abstract = {Understanding ion transport mechanisms in plants is crucial for enhancing stress tolerance and crop yields. The Arabidopsis thaliana High-Affinity Potassium Transporter 1; 1 (AtHKT1; 1) is vital for maintaining sodium and potassium balance under saline stress. However, the structural and functional effects of genetic variants in AtHKT1; 1, especially in potassium chloride (KCl) environments, are not fully understood. This research combiness Genome-Wide Association Studies (GWAS) with CRISPR-based functional validation to examine AtHKT1; 1 gene variants, focusing on the protein structure with PDB ID: 8W9O. The study first used GWAS and SNP discovery to find significant genetic variations linked to ion transport and salt tolerance. Candidate SNPs were selected based on their statistical significance and potential biological roles. Structural analysis of the protein 8W9O involved PDB and MMDB resources, with validation through ERRAT and visualization/mutation mapping in PyMOL. InterProScan was used to identify conserved functional motifs. To validate SNP effects, CRISPR guide RNAs were designed with E-CRISP and CHOPCHOP, targeting key gene regions for precise editing. This integrated approach linked genetic variations to structural changes and their potential impact on ion binding and transport, especially under KCl conditions. Results showed certain SNPs cause conformational shifts in key transmembrane regions of AtHKT1; 1, possibly influencing ion selectivity and transport efficiency. Structural validation confirmed the accuracy of the modeled variants, and domain analysis revealed disruptions in conserved functional areas. CRISPR strategies proved feasible for precise gene editing to test these functional hypotheses. Overall, this study offers a comprehensive framework that combines GWAS, structural bioinformatics, and CRISPR technology to explore how genetic variants affect AtHKT1; 1 function. These insights improve understanding of ion transport regulation in saline environments and support the development of genetically modified crops with enhanced salt tolerance.},
year = {2026}
}
TY - JOUR T1 - Integrative GWAS and CRISPR Functional Validation of Gene Variant Structure of AtHKT1; 1 in KCl AU - Shephali Sachan AU - Uma Kumari Y1 - 2026/04/25 PY - 2026 N1 - https://doi.org/10.11648/j.cbb.20261401.13 DO - 10.11648/j.cbb.20261401.13 T2 - Computational Biology and Bioinformatics JF - Computational Biology and Bioinformatics JO - Computational Biology and Bioinformatics SP - 26 EP - 40 PB - Science Publishing Group SN - 2330-8281 UR - https://doi.org/10.11648/j.cbb.20261401.13 AB - Understanding ion transport mechanisms in plants is crucial for enhancing stress tolerance and crop yields. The Arabidopsis thaliana High-Affinity Potassium Transporter 1; 1 (AtHKT1; 1) is vital for maintaining sodium and potassium balance under saline stress. However, the structural and functional effects of genetic variants in AtHKT1; 1, especially in potassium chloride (KCl) environments, are not fully understood. This research combiness Genome-Wide Association Studies (GWAS) with CRISPR-based functional validation to examine AtHKT1; 1 gene variants, focusing on the protein structure with PDB ID: 8W9O. The study first used GWAS and SNP discovery to find significant genetic variations linked to ion transport and salt tolerance. Candidate SNPs were selected based on their statistical significance and potential biological roles. Structural analysis of the protein 8W9O involved PDB and MMDB resources, with validation through ERRAT and visualization/mutation mapping in PyMOL. InterProScan was used to identify conserved functional motifs. To validate SNP effects, CRISPR guide RNAs were designed with E-CRISP and CHOPCHOP, targeting key gene regions for precise editing. This integrated approach linked genetic variations to structural changes and their potential impact on ion binding and transport, especially under KCl conditions. Results showed certain SNPs cause conformational shifts in key transmembrane regions of AtHKT1; 1, possibly influencing ion selectivity and transport efficiency. Structural validation confirmed the accuracy of the modeled variants, and domain analysis revealed disruptions in conserved functional areas. CRISPR strategies proved feasible for precise gene editing to test these functional hypotheses. Overall, this study offers a comprehensive framework that combines GWAS, structural bioinformatics, and CRISPR technology to explore how genetic variants affect AtHKT1; 1 function. These insights improve understanding of ion transport regulation in saline environments and support the development of genetically modified crops with enhanced salt tolerance. VL - 14 IS - 1 ER -
Department of Bioinformatics, Bioinformatics Project and Research Institute, Noida, India
Department of Bioinformatics, Bioinformatics Project and Research Institute, Noida, India
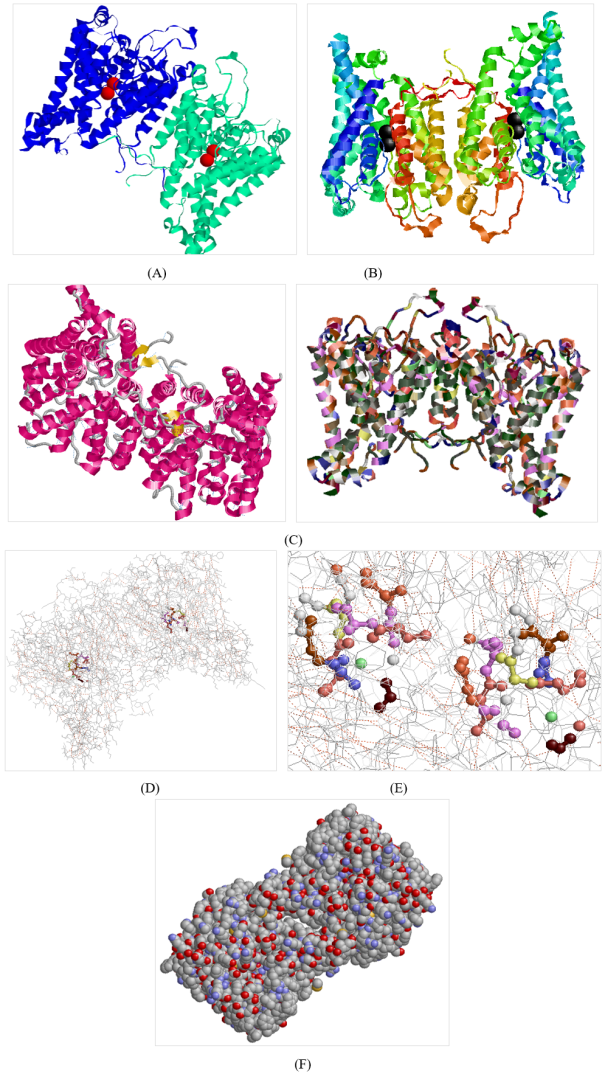
Figure 1. 8W9O 3-D structure visualization by Raswin (A) Displays two protein units (A in blue, B in green), each centered with 2 potassium (K+) ions in red. (B) Highlights termini—N-termini (blue) and C-termini (red)—along with varied colors indicating different helix chains (C) Shows the complex secondary and tertiary structures, including alpha-helices and beta-sheets (helix - magenta, sheets - yellow, and turn - grey) (D) Reveals the amino acid composition, with Leucine (olive green) as the most abundant residue (976 atoms), and Cysteine as the least (96 atoms) (E) Illustrates the active site residues within 5A0 distance from ligand, dominated by light purple (Valine) and light salmon (Asparagine) color (F) Displays the surface atoms with standard atomic coloring: Carbon (grey), Nitrogen (blue), Oxygen (red), Sulfur (yellow).
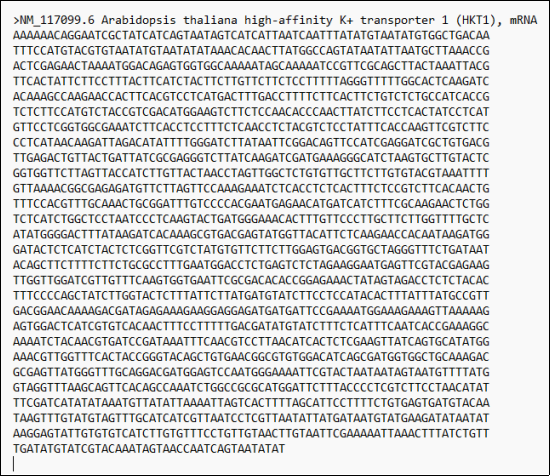
Figure 2. Presenting AtHKT1; 1 amino acid sequences (8W9O) with protein chain A and B separately and mRNA sequence (NM_117099.6).
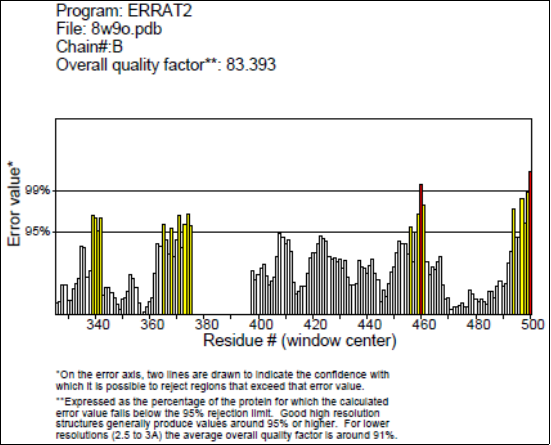
Figure 3. ERRAT structural validation of AtHKT1; 1 (8W9O) protein.
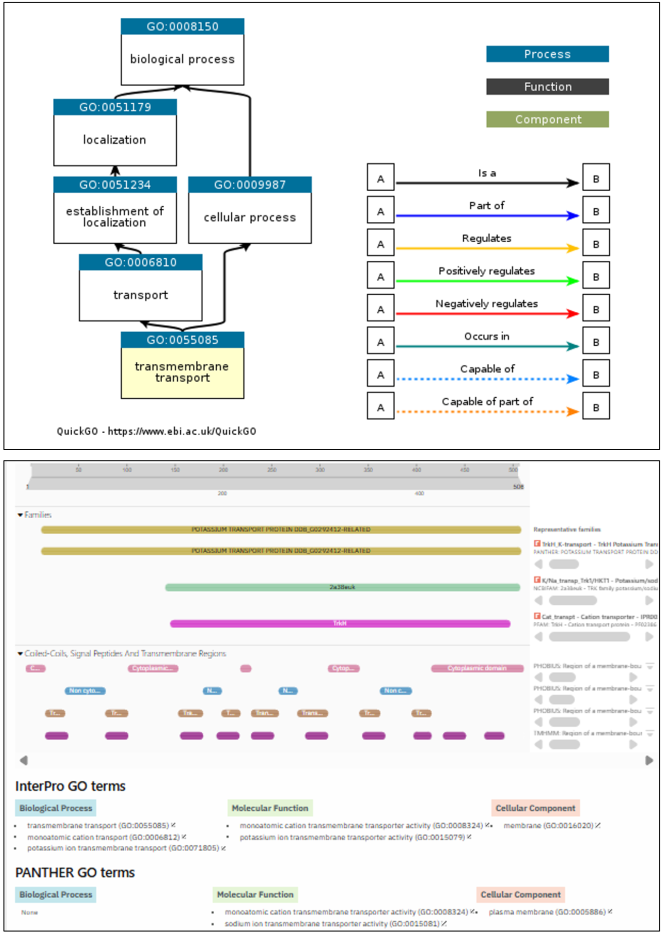
Figure 4. Functional validation of AtHKT1; 1 (8W9O) protein by InterProScan.
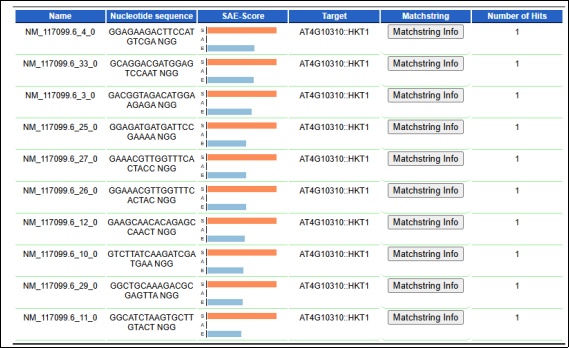
Figure 5. CRISPR/Cas9 guide RNA (gRNA) candidates targeting AtHKT1; 1 (AT4G10310) identified through in silico analysis. Each row represents a unique gRNA sequence aligned to the AtHKT1; 1 coding region, along with corresponding specificity, activity, and efficiency (SAE) scores. All designed gRNAs show a single on-target hit within the AtHKT1; 1 locus, indicating high target specificity suitable for functional validation of allelic variants associated with salt tolerance in Arabidopsis thaliana.
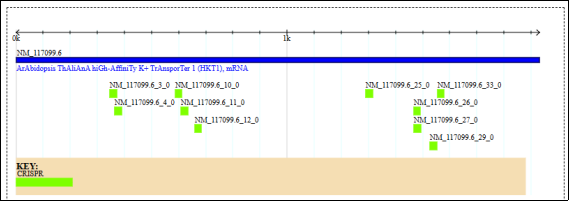
Figure 6. Graphical representation of CRISPR/Cas9 target sites designed within the AtHKT1; 1 (AT4G10310) gene of Arabidopsis thaliana. The blue bar denotes the AtHKT1; 1 mRNA sequence (accession: NM_117099.6), while green boxes indicate predicted CRISPR guide RNA (gRNA) binding positions along the coding region. Each labeled gRNA corresponds to specific target sites identified in silico, distributed across the 1 kb gene region. This map illustrates the spatial organization of selected high-specificity gRNAs for functional genome-editing validation of AtHKT1; 1 variants associated with salt tolerance.
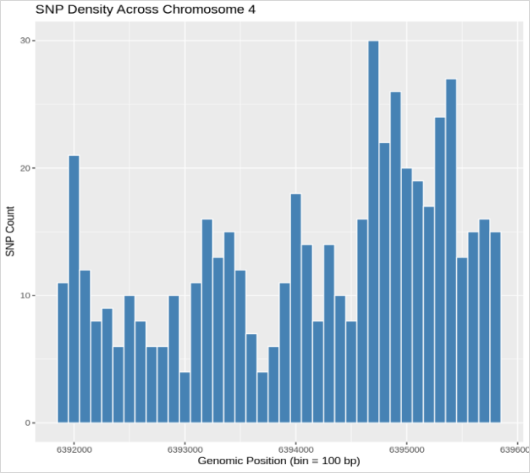
Figure 7. SNP density plot across the AtHKT1; 1 gene region on chromosome 4 (positions 6,391,800–6,395,900 bp) in Arabidopsis thaliana. Each bar represents the number of single-nucleotide polymorphisms (SNPs) per 100 bp window, illustrating uneven variant distribution with higher SNP clustering toward the 3′ end of the gene, indicative of localized mutational hotspots potentially associated with allelic diversity in salt tolerance.
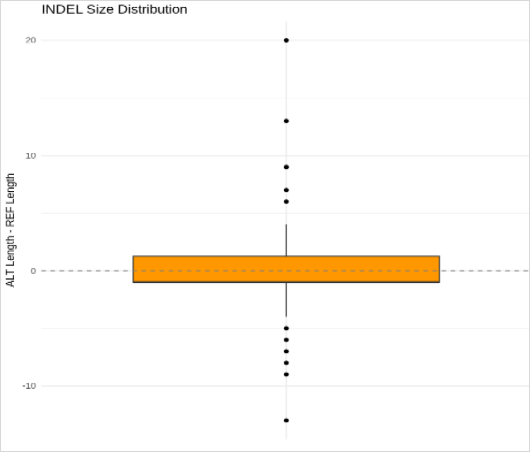
Figure 8. Box plot showing INDEL size distribution in the AtHKT1; 1 gene region. The Y-axis represents the difference between ALT and REF sequence lengths (ALT–REF), where positive values indicate insertions and negative values indicate deletions. Most INDELs are small (±3 bp) with a median near zero, reflecting balanced insertion–deletion rates, while a few outliers represent rare larger events (~–12 bp).
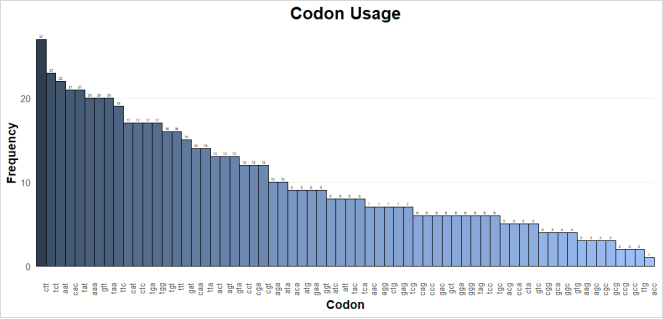
Figure 9. Bar chart showing the frequency of codon usage in AtHKT1; 1 sequence.
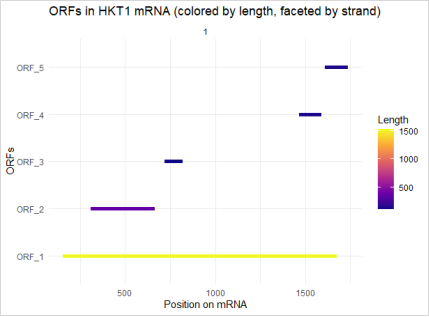
Figure 10. Representing open reading frames detected in the HKT1 gene sequence. Y axis = ORF1 TO ORF_5, x-axix = Position on the DNA (Nucleotide coordinate), Color Gradient = Represents length of ORF (from short =dark blue to long=yellow).
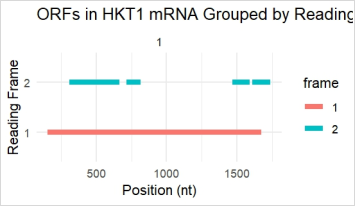
Figure 11. Representing open reading frames detected in the HKT1 gene sequence. Y-axis (Reading frame) = Frame 1 (Red) is a single long continuous ORF, representing dominant long ORF from ~250 to ~1600nt, true coding sequence for HKT1 protein. Frame 2 = blue Multiple shorter ORF Scattered through sequence, might be non-coding, regulatory, or non-functional peptide.
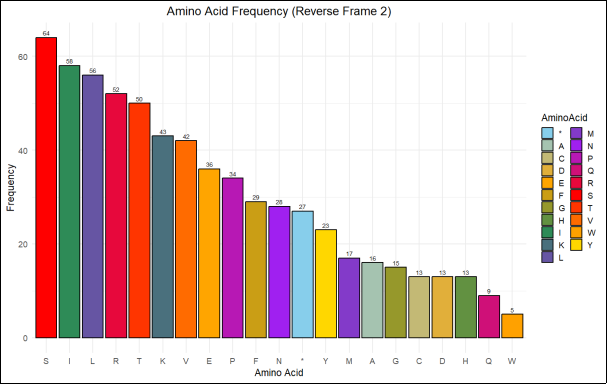
Figure 12. Bar chart illustrating the amino acid frequency distribution in Reverse Frame 2 (RF2) of the AtHKT1; 1 sequence. The X-axis represents amino acids (single-letter codes), while the Y-axis indicates their frequency of occurrence. Distinct colors differentiate individual amino acids for visual clarity. The most abundant residues are leucine (L, 76), serine (S, 62), isoleucine (I, 57), and valine (V, 50), reflecting a predominance of hydrophobic and polar amino acids typically associated with transmembrane helices in ion transporter proteins.
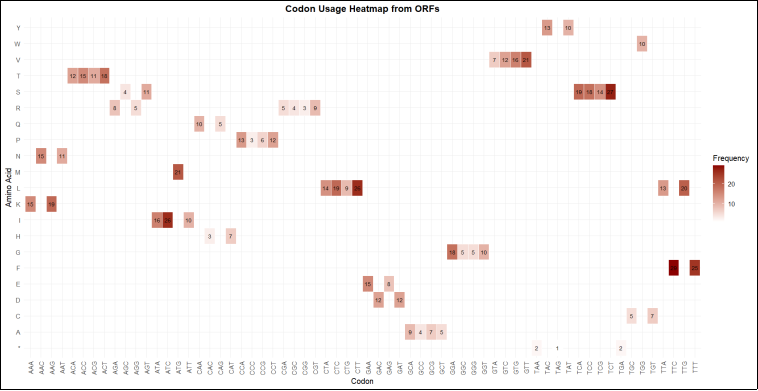
Figure 13. Codon Usage heatmap displaying frequency of codon usage for respective amino acid. Y-axis = Amino acid with possibly one letter code, X-axis = Codon (Triplet sequence gaa, tgc) Color scale gradient (red) = indicate frequency of codon usage, Darker shades/red = higher frequency (dark red =high frequency =more use), lighter = lower usage (Less rare Usage). Each square = one codon for specific amino acid.

Figure 14. Horizontal bar graph illustrates AtHKT1; 1 gene structure including both strands translated results.
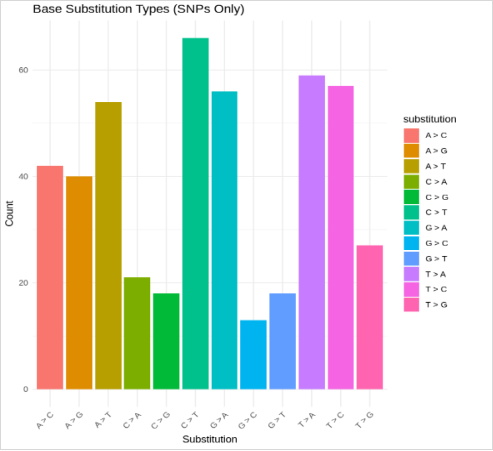
Figure 15. Bar graph describes SNP created by base substitution with major substitutions shown by: C > G, G > A, T > A, and T > C, dominated by transversion type.
Information

