This research aimed to understand the effects induced by PP, PQ and QQ complexes on the geometry, thermodynamic stability and vibration frequencies of salicylideneamines. It planned to determine the interactions within them. It envisaged identifying the impact of the solvent during the creation of these dimers by analyzing variations in electronic energies and associated dipole moments. To do this, the study utilized exploited DFT combined with sets of basis functions such as those of Pople and the HF method to optimize the geometries of P or Q monomers and PP, PQ or QQ dimers. The results obtained were employed for the calculations of NBO and QTAIM in the case of isopropyl amine N-(2,3-dihydroxybenzylidene) structures. For the last objective, SPSS Statistics v27 software was used to compare variations in energies or electronic moments during transitions from monomers to dimers. This methodological approach made it possible to prove that the electronic transitions σ → σ* and π → π* improve the equilibrium of the P and Q monomers. Their “dimeric” associations are steadied by those of the Lp → σ* and Lp → π* type with the heteroatoms of the PP, PQ and QQ complexes. These phenomena are obtained thanks to the hydrogen bonds established between the latter and the hydrogen favourable to these interactions. The observed “thermochromic” and photochromic trends can be explained by the stability of the PQ. The presence of the thermodynamically disadvantaged Q tautomer is also justified. The nature and polarity of the solvents don’t significantly influence these latter results.
| Published in | International Journal of Computational and Theoretical Chemistry (Volume 13, Issue 1) |
| DOI | 10.11648/j.ijctc.20251301.12 |
| Page(s) | 13-24 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Salicylideneamines, Interactions, Quantum Calculations, Tautomers, NBO, QTAIM
Monohydroxylated salicylideneamines | ||
|---|---|---|
Tautomeric equilibrium |
| |
acronyms | Radical R | Molecular structure name |
A |
| N-(2-hydroxybenzylidene) benzylamine |
B |
| N-(2-hydroxybenzylidene)-2-chlorobenzylamine |
C |
| N-(2-hydroxybenzylidene) cyclopropylamine |
D |
| N-(2-hydroxybenzylidene) paramethyl benzylamine |
E |
| N-(2-hydroxybenzylidene) isopropylamine |
Dihydroxylated salicylideneamines | ||
| ||
Radical |
| N-(2,3- dihydroxybenzylidene) isopropylamine |
Parameters | Dimer (PP) | Dimer (PQ) | Dimer (QQ) |
|---|---|---|---|
Electronic energies E (au) | -1188.531392 | -1188.531530 | -1188.530701 |
Dipole moments (D) | 2.2139 | 3.7422 | 0.00028 |
Geometric parameters of HB | |||
O33 (Q)… H-O14 (P) | 1.890 | 1.764 | 1.840 |
O16 (P)… H-O31 (Q) | 1.890 | 1.960 | 1.840 |
<O33(Q)…H-O14(P) | 159.58 | 155.51 | 147.37 |
<O16(P)…H-O31(Q) | 159.58 | 152.06 | 147.37 |
Pseudo-cycle | |||
N… HO/O… NH | 1.683 | 1.68979 (P)/1.738 (Q) | 1.726 |
<O33(Q)…H-O14(P) | 148.54 | 148.54 (P)/137.48 (Q) | 137.63 |
Null Hypothesis | Sig.a | Decision |
|---|---|---|
The distribution of E is normal with mean 4.705 and standard deviation 1.122787. | 0.184 | Retain the null hypothesis. |
The distribution of is normal with mean 1.974 and standard deviation .413118. | 0.967 | Retain the null hypothesis. |
Tests | E | |
|---|---|---|
Kaiser-Meyer-Olkin Measure of Sampling Adequacy. | 0.500 | 0.500 |
Bartlett’s Test of Sphericity | ||
Approx. Chi-Square | 4.008 | 2.025 |
Df | 1 | 1 |
Sig. | 0.045 | 0.155 |
Variables | Levene Statistic | df1 | df2 | Sig. | |
|---|---|---|---|---|---|
E | Based on Mean | 0.012 | 2 | 12 | 0.988 |
| Based on Mean | 0.011 | 2 | 12 | 0.989 |
Molecules | Gas phase | Cyclohexane | CCl4 | Median |
|---|---|---|---|---|
A | 4.936 | 3.827 | 3.683 | 3.827 |
B | 5.965 | 5.016 | 4.889 | 5.016 |
C | 7.174 | 5.737 | 5.557 | 5.737 |
D | 4.910 | 3.774 | 3.627 | 3.774 |
E | 4.915 | 3.381 | 3.186 | 3.381 |
Molecules | Gas phase | Cyclohexane | CCl4 | Median |
|---|---|---|---|---|
A | 1.468 | 1.882 | 1.925 | 1.882 |
B | 1.212 | 1.556 | 1.600 | 1.556 |
C | 1.936 | 2.361 | 2.417 | 2.361 |
D | 1.680 | 2.110 | 2.168 | 2.110 |
E | 2.154 | 2.540 | 2.594 | 2.540 |
Total N | 15 |
|---|---|
Test Statistic | 4.340a,b |
Degree of Freedom | 2 |
Asymptotic Sig.(2-sided test) | 0.114 |
Structures | Gas phase | Cyclohexane | CCl4 | Median |
|---|---|---|---|---|
A | 1.468 | 1.882 | 1.925 | 1.882 |
B | 1.212 | 1.556 | 1.600 | 1.556 |
C | 1.936 | 2.361 | 2.417 | 2.361 |
D | 1.680 | 2.110 | 2.168 | 2.110 |
E | 2.154 | 2.540 | 2.594 | 2.540 |
Total N | 15 |
|---|---|
Test Statistic | 3.120a,b |
Degree of Freedom | 2 |
Asymptotic Sig.(2-sided test) | 0.21 |
Electronic transitions | E(2) (kcal.mol-1) | CT (me) | |
|---|---|---|---|
π(C1-C2) | π*(C3-C4) | 37,57 | 123,10 |
π(C1-C2) | π*(C5-C6) | 33,46 | 113,81 |
π(C3-C4) | π*(C1-C2) | 39,58 | 133,47 |
π(C3-C4) | π*(C5-C6) | 46,62 | 162,57 |
π(C5-C6) | π*(C1-C2) | 40,43 | 134,35 |
π(C5-C6) | π*(C3-C4) | 31,99 | 103,97 |
π(C5-C6) | π*(C10-N12) | 29,01 | 105,80 |
π(C18-C23) | π*(C19-C20) | 35,25 | 120,87 |
π(C18-C23) | π*(C27-N29) | 31,95 | 113,29 |
π(C19-C20) | π*(C18-C23) | 48,04 | 180,00 |
Electronic transitions | E(2) (kcal.mol-1) | CT (me) | Environment | Fragment | |
|---|---|---|---|---|---|
Lp(1)N12 | *(C10-H11) | 13,15 | 16,73 | Intramolecular | P |
Lp(1)N12 | *(O16-H17) | 45,26 | 56,24 | Pseudo-cycle | P |
Lp(1)O14 | *(C4-C5) | 10,96 | 11,11 | Intramolecular | P |
Lp(1)O16 | *(C5-C6) | 9,54 | 9,45 | Intramolecular | P |
Lp(1)O16 | *(O31-H32) | 7,12 | 7,32 | Intermolecular | P-Q |
Lp(1)O16 | *(O31-H32) | 7,97 | 10,43 | Intermolecular | P-Q |
Lp(1)O33 | *(O14-H15) | 15,73 | 14,61 | Intermolecular | Q-P |
Lp(3)O33 | *(O14-H15) | 9,69 | 15,43 | Intermolecular | Q-P |
Lp(1)O33 | *(N29-H34) | 9,43 | 9.1 | Pseudo-cycle | Q |
Lp(2)O33 | *(C21-C22) | 19,07 | 26,07 | Intramolecular | Q |
Lp(2)O33 | *(C22-C23) | 14,39 | 19,67 | Intramolecular | Q |
Lp(2)O33 | *(N29-H34) | 23,75 | 33,44 | Pseudo-cycle | Q |
Lp(1)O31 | *(C21-C22) | 10,08 | 10,58 | Intramolecular | Q |
Lp(2)O31 | *(C20-C21) | 38,53 | 98,91 | Intramolecular | Q |
Lp(1)C6 | *(C1-C2) | 104,29 | 849,82 | Intramolecular | P |
Lp(1)C6 | *(C10-N12) | 66,40 | 612,88 | Intramolecular | P |
Lp(1)C23 | *(C18-C19) | 76,91 | 512,58 | Intramolecular | Q |
Lp(1)C23 | *(C27-N29) | 205,70 | 1910,12 | Intramolecular | Q |
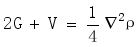 with local energy density
with local energy density 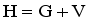 .
. PP | Phenolic-Phenolic |
PQ | Phenolic-Quinonic |
Quinonic-Quinonic | |
HF | Hatree-Fock |
DFT | Density Functional Theory |
NBO | Natural Bond Orbitals |
QTAIM | Quantum Theory of Atoms in the Molecule |
| [1] | Bhagat, S., Sharma, N., Chundawat, T. Singh. Synthesis of some Salicylaldehyde-based Schiff bases in Aqueous Media, Journal of Chemistry, 2013, 1-4. |
| [2] | Harada, J., Uekusa, H., Ohashi, Y. X-ray Analysis of Structural Changes in Photochromic Salicylideneaniline Crystals. Solid-state Reaction Induced by Two-photon Excitation, Journal of the American Chemical Society. 1999, 121(24), 5809-5810. |
| [3] | Margerum, J. D., Miller, L. S., Saito, E., Brown, M. S., Mosher, H. S., Hardwick, R. Phototropism of Ortho-nitroBenzyl Derivatives, Journal of Physical Chemistry. 1962, 66(12), 2434-2438. |
| [4] | Loy, M., Huffman, R. K., Ullman, E. F. Photoenolization of Some Photochromic Ketones. The Scope and Mechanism of the Reaction, Journal of the American Chemical Society. 1965, 87(23), 5417-5423. |
| [5] | Taneda, M., Koyama, H., Kawato. T. Vapour Switching of Photochromism of Methylenebis {N- (3,5 di-tert-butylsalicylidene) Aniline} Crystals, Chemistry Letters. 2007, 36(3), 354-355. |
| [6] | Ünver, H., Kabak, M., Zengin, D. M., Durlu, T. N. Keto-enol tautomerism, conformations, and structure of 1- [N- (4- chlorophenyl)] aminomethylidene-2 (1H) naphthalenone, Journal of chemical crystallography. 2001, 31(4), 203-209. |
| [7] | Mikami, M. Nakamura, S. First-principle Study of Salicylideneaniline Molecular Crystals: Tautomerization Reaction Involving Intermolecular Hydrogen Bonds, Physical. Review B. 2004, 69(13), 1-8. |
| [8] | Desiraju, G. R. Chemistry beyond the Molecule, Nature. 2001, 412(6845), 397-400. |
| [9] | Ostertagova, E., Ostertag, O., Kovac, Applied mechanics and materials, J. Methodology and Application of the Kruskal-Wallis Test. 2014, 611, 115-120. |
| [10] | Chan, Y., Walmsley, R. P. Learning and Understanding the Kruskal-Wallis One-way Analysis-of-variance-by-rank Test for Differences among Three or More Independent Groups, Physical therapy. 1997, 77(12). |
| [11] | Lilliefors, H. W. On the Kolmogorov-Smirnov Test for Normality with Mean and Variance Unknown. Journal of the American Statistical Association. 1967, 62(318), 399-402. |
| [12] | Pallant, J. SPSS Survival Manual: A Step-by-step Guide to Data Analysis Using IBM SPSS.7th Edition, London: Routledge, 2023, 1-378. |
| [13] | Laflamme, S., Zhou, R. — M. Méthodes statistiques en sciences humaines. 2de Édition, Sudbury: Prise de parole. 2014, 1-61. |
| [14] | Parresol, B. R. Baldcypress. Height—diameter Equations and their Prediction Confidence Intervals, Canadian journal of forest research. 1992, 22(9), 1429-1434. |
| [15] | Weinhold, F., Landis, C. R., Glendening, E. D. What is NBO Analysis and How is it Useful? International Reviews in Physical Chemistry. 2016, 35(3), 399-440. |
| [16] | Rozas, I., Alkorta, I., Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors, Journal of the American Chemical Society. 2000, 122(45), 11154-11161. |
| [17] | Abramov, Y. A. On the Possibility of Kinetic Energy Density Evaluation from the Experimental Electron-Density Distribution, Acta Crystallographica Section A Foundations of Crystallography. 1997, 53(3), 264-272. |
| [18] | Ziółkowski, M., Grabowski, S. J., Leszczynski, J. Cooperativity in Hydrogen-Bonded Interactions: Ab initio and “Atoms in Molecules” Analyses, The Journal of Physical Chemistry A. 2006, 110(20), 6514-6521. |
APA Style
N’guessan, B. R., Essoh, A. E., Adenidji, G., Bamba, E. H. S. (2025). Highlighting of Properties of Thermochromy and Photochromy in Salicylideneamines. International Journal of Computational and Theoretical Chemistry, 13(1), 13-24. https://doi.org/10.11648/j.ijctc.20251301.12
ACS Style
N’guessan, B. R.; Essoh, A. E.; Adenidji, G.; Bamba, E. H. S. Highlighting of Properties of Thermochromy and Photochromy in Salicylideneamines. Int. J. Comput. Theor. Chem. 2025, 13(1), 13-24. doi: 10.11648/j.ijctc.20251301.12
@article{10.11648/j.ijctc.20251301.12,
author = {Boka Robert N’guessan and Akpa Eugène Essoh and Ganiyou Adenidji and El Hadji Sawaliho Bamba},
title = {Highlighting of Properties of Thermochromy and Photochromy in Salicylideneamines
},
journal = {International Journal of Computational and Theoretical Chemistry},
volume = {13},
number = {1},
pages = {13-24},
doi = {10.11648/j.ijctc.20251301.12},
url = {https://doi.org/10.11648/j.ijctc.20251301.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijctc.20251301.12},
abstract = {This research aimed to understand the effects induced by PP, PQ and QQ complexes on the geometry, thermodynamic stability and vibration frequencies of salicylideneamines. It planned to determine the interactions within them. It envisaged identifying the impact of the solvent during the creation of these dimers by analyzing variations in electronic energies and associated dipole moments. To do this, the study utilized exploited DFT combined with sets of basis functions such as those of Pople and the HF method to optimize the geometries of P or Q monomers and PP, PQ or QQ dimers. The results obtained were employed for the calculations of NBO and QTAIM in the case of isopropyl amine N-(2,3-dihydroxybenzylidene) structures. For the last objective, SPSS Statistics v27 software was used to compare variations in energies or electronic moments during transitions from monomers to dimers. This methodological approach made it possible to prove that the electronic transitions σ → σ* and π → π* improve the equilibrium of the P and Q monomers. Their “dimeric” associations are steadied by those of the Lp → σ* and Lp → π* type with the heteroatoms of the PP, PQ and QQ complexes. These phenomena are obtained thanks to the hydrogen bonds established between the latter and the hydrogen favourable to these interactions. The observed “thermochromic” and photochromic trends can be explained by the stability of the PQ. The presence of the thermodynamically disadvantaged Q tautomer is also justified. The nature and polarity of the solvents don’t significantly influence these latter results.
},
year = {2025}
}
TY - JOUR T1 - Highlighting of Properties of Thermochromy and Photochromy in Salicylideneamines AU - Boka Robert N’guessan AU - Akpa Eugène Essoh AU - Ganiyou Adenidji AU - El Hadji Sawaliho Bamba Y1 - 2025/03/07 PY - 2025 N1 - https://doi.org/10.11648/j.ijctc.20251301.12 DO - 10.11648/j.ijctc.20251301.12 T2 - International Journal of Computational and Theoretical Chemistry JF - International Journal of Computational and Theoretical Chemistry JO - International Journal of Computational and Theoretical Chemistry SP - 13 EP - 24 PB - Science Publishing Group SN - 2376-7308 UR - https://doi.org/10.11648/j.ijctc.20251301.12 AB - This research aimed to understand the effects induced by PP, PQ and QQ complexes on the geometry, thermodynamic stability and vibration frequencies of salicylideneamines. It planned to determine the interactions within them. It envisaged identifying the impact of the solvent during the creation of these dimers by analyzing variations in electronic energies and associated dipole moments. To do this, the study utilized exploited DFT combined with sets of basis functions such as those of Pople and the HF method to optimize the geometries of P or Q monomers and PP, PQ or QQ dimers. The results obtained were employed for the calculations of NBO and QTAIM in the case of isopropyl amine N-(2,3-dihydroxybenzylidene) structures. For the last objective, SPSS Statistics v27 software was used to compare variations in energies or electronic moments during transitions from monomers to dimers. This methodological approach made it possible to prove that the electronic transitions σ → σ* and π → π* improve the equilibrium of the P and Q monomers. Their “dimeric” associations are steadied by those of the Lp → σ* and Lp → π* type with the heteroatoms of the PP, PQ and QQ complexes. These phenomena are obtained thanks to the hydrogen bonds established between the latter and the hydrogen favourable to these interactions. The observed “thermochromic” and photochromic trends can be explained by the stability of the PQ. The presence of the thermodynamically disadvantaged Q tautomer is also justified. The nature and polarity of the solvents don’t significantly influence these latter results. VL - 13 IS - 1 ER -
Constitution and Reaction of Matter Laboratory, Training and Research Unit in Structural, Material and Technological Sciences, Felix Houphouet-Boigny University, Abidjan, Côte d’Ivoire
Constitution and Reaction of Matter Laboratory, Training and Research Unit in Structural, Material and Technological Sciences, Felix Houphouet-Boigny University, Abidjan, Côte d’Ivoire
Constitution and Reaction of Matter Laboratory, Training and Research Unit in Structural, Material and Technological Sciences, Felix Houphouet-Boigny University, Abidjan, Côte d’Ivoire; Sciences and Technologies of Environment Laboratory, Training and Research Unit in Environment, Jean Lorougnon Guede University, Daloa, Côte d’Ivoire
Constitution and Reaction of Matter Laboratory, Training and Research Unit in Structural, Material and Technological Sciences, Felix Houphouet-Boigny University, Abidjan, Côte d’Ivoire

Figure 1. Tautomeric equilibrium in salicylidenamines.
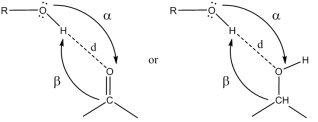
Figure 2. Geometric Parameters of Hydrogen Bond.
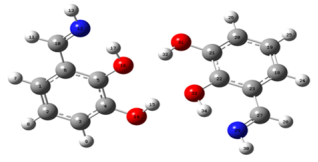
Figure 3. PP Complex.
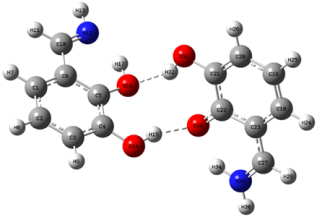
Figure 4. PQ Complex.
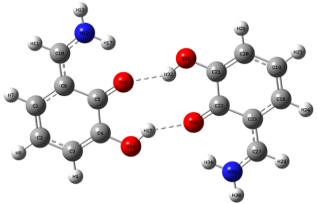
Figure 5. QQ Complex.
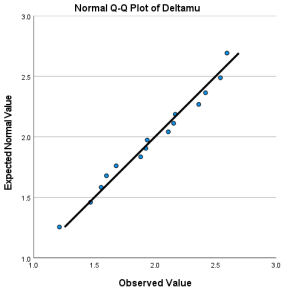
Figure 6. Q-Q diagram associated with .
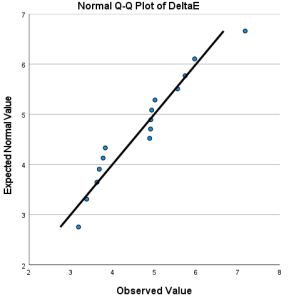
Figure 7. Q-Q diagram associated with ∆E.
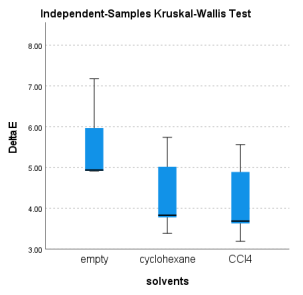
Figure 8. Simple Box of the Energy Variations (kcal.mol-1) by the Solvents.
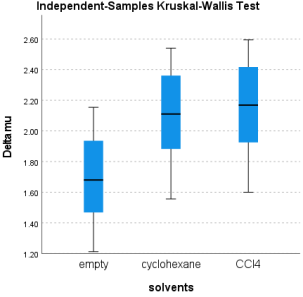
Figure 9. Simple Box of the Dipole Moments Variations (D) by the Solvents.
Information









