Abstract
Hypophosphatasia (HPP), a rare metabolic disorder caused by mutations in the ALPL gene, leads to deficient alkaline phosphatase activity and presents unique clinical challenges for adult patients, including fractures, chronic pain, and dental issues. This disorder is often underdiagnosed due to its variable clinical presentation and overlap with other conditions, further complicating timely intervention. Early diagnosis is critical for effective management; however, current diagnostic criteria have limitations, often resulting in delays. Insights from the Global HPP Registry reveal significant disease burden and treatment gaps in treated and untreated adults. These insights underscore the importance of multidisciplinary approaches in addressing HPP-related complications, including musculoskeletal and systemic manifestations. Enzyme replacement therapy (ERT) with asfotase alfa has proven effective in reducing disease burden and improving quality of life. Recent clinical evidence suggests that ERT not only alleviates symptoms but may also prevent disease progression when initiated early. Emerging therapies and alternative regimens like teriparatide dosing adjustments are being explored for their potential benefits. In addition, advanced imaging modalities and biomarker studies are improving diagnostic accuracy and monitoring of therapeutic outcomes. This comprehensive review highlights the significant challenges and advances in diagnosing and treating HPP in adults. Ongoing research aims to enhance diagnosis and treatment through genetic testing and personalized medicine, focusing on identifying and addressing knowledge gaps to improve care for adult HPP patients. Collaborative efforts between researchers, clinicians, and patient advocacy groups are crucial for driving innovation and improving access to care. Continued research and innovation are essential, and healthcare professionals must stay informed about the latest advancements in HPP diagnosis and treatment to ensure optimal patient care. By addressing these challenges, the field can move closer to improving the lives of adults living with HPP.
Keywords
Hypophosphatasia, Mineralization, Osteomalacia, Enzyme Replacement Therapy
1. Introduction
Hypophosphatasia (HPP) is a rare, inherited metabolic disorder characterized by defective mineralization of bones and teeth due to mutations in the ALPL gene encoding the tissue non-specific alkaline phosphatase (TNSALP) enzyme. This enzyme plays a critical role in bone mineralization by breaking down inorganic pyrophosphate, a natural inhibitor of bone formation. When TNSALP is deficient or dysfunctional, inorganic pyrophosphate accumulates, leading to improper bone mineralization and a spectrum of clinical manifestations ranging from perinatal lethality to mild, late-onset forms in adults
| [1] | Simon, S.; Resch, H.; Klaushofer, K.; Roschger, P.; Zwerina, J.; Kocijan, R. Hypophosphatasia: From Diagnosis to Treatment. Curr Rheumatol Rep. 2018, 20, 69. https://doi.org/10.1007/s11926-018-0778-5 |
| [2] | Tournis, S.; Yavropoulou, M. P.; Polyzos, S. A.; Doulgeraki, A. Hypophosphatasia. J. Clin. Med. 2021, 10, 5676. https://doi.org/10.3390/jcm10235676 |
[1, 2]
.
The diagnosis and treatment of hypophosphatasia in adults present unique challenges and are of significant clinical importance for several reasons. Adult-onset HPP is often underdiagnosed or misdiagnosed due to its variable presentation and overlap with more common conditions such as osteoporosis, osteoarthritis, and other metabolic bone diseases. This can lead to inappropriate treatments that may exacerbate the condition, such as bisphosphonates, contraindicated in HPP. Therefore, raising awareness and improving diagnostic criteria for adult-onset HPP is crucial for ensuring patients receive accurate diagnoses and appropriate management
| [3] | Martos-Moreno, G. Á.; Calzada, J.; Couce, M. L.; Argente, J. Hypophosphatasia: Clinical manifestations, diagnostic recommendations and therapeutic options. An Pediatría. 2018, 88, 356. e1-356. e11. https://doi.org/10.1016/j.anpede.2017.06.006 |
| [4] | Mornet, E.; Taillandier, A.; Domingues, C.; Dufour, A.; Benaloun. E.; et al. Hypophosphatasia: a genetic-based nosology and new insights in genotype-phenotype correlation. Eur J Hum Genet. 2021, 29, 289–299. https://doi.org/10.1038/s41431-020-00732-6 |
[3, 4]
.
Globally, the prevalence of HPP is estimated to range from 1 in 100,000 to 1 in 300,000 live births, with the adult form being less frequently recognized but not necessarily less common. The exact number of adults living with HPP remains uncertain due to underdiagnosis and misclassification of the disease. However, it is estimated that thousands of adults worldwide suffer from this condition, potentially more, given the increasing recognition of the milder phenotypes
| [5] | Seefried, L.; Dahir, K.; Petryk, A.; Högler, W.; Linglart, A.; et al. Burden of Illness in Adults With Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Bone Miner Res. 2020, 35, 2171–2178. https://doi.org/10.1002/jbmr.4130 |
| [6] | Kishnani, P.; Petryk, A.; Hoegler, W.; Linglart, A.; et al. SUN-529 Burden of Illness in Adults with Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Endocr Soc. 2019, 3, SUN-529. https://doi.org/10.1210/js.2019-SUN-529 |
| [7] | González-Cejudo, T.; Villa-Suárez, J. M.; Ferrer-Millán, M.; Andújar-Vera, F.; et al. Mild hypophosphatasia may be twice as prevalent as previously estimated: an effective clinical algorithm to detect undiagnosed cases. Clin Chem Lab Med. 2024, 62, 128–137. https://doi.org/10.1515/cclm-2023-0427 |
[5-7]
.
In recent years, significant advances have been made in understanding the genetic and molecular mechanisms underlying HPP, leading to the development of novel therapeutic approaches. One of the most notable advancements is introducing ERT with asfotase alfa, a recombinant human TNSALP, which has shown promise in improving clinical outcomes in pediatric and adult patients with HPP
.
Given the clinical complexities, the potential for misdiagnosis, and the advances in therapeutic options, it is imperative to comprehensively review the challenges and recent developments in diagnosing and treating hypophosphatasia in adults. This review aims to provide a detailed overview of the current understanding of HPP, discuss the difficulties encountered in clinical practice, and highlight the latest advances in diagnostic and therapeutic strategies, ultimately guiding better clinical decision-making and improving patient outcomes. Ultimately, this review aims to enhance patient outcomes and quality of life by promoting informed clinical decision-making and identifying areas for future research.
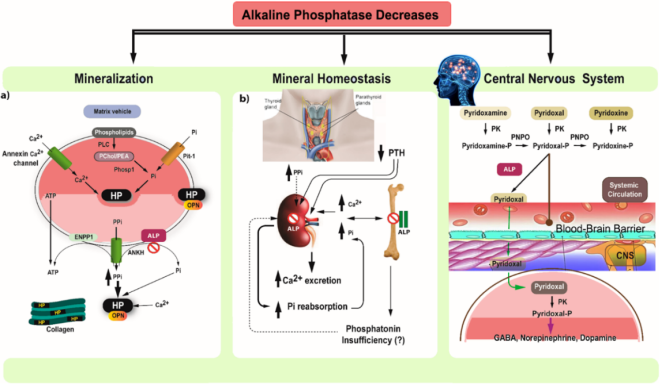
Figure 1. Key processes in mineralization and HPP.
(a) Primary Mineralization and Matrix Vesicles (MV): Mineralization begins in matrix vesicles (MV), which are osteoblast membrane buds. Inorganic phosphate (Pi) is produced within the MV by PHOSPHO-1 acting on PChol and PEA, transported by Pit-1. Pi combines with calcium to form hydroxyapatite crystals, causing MV rupture and further mineralization in the extracellular matrix on collagen fibrils. Pyrophosphate (PPi) inhibits mineralization, while TNAP promotes it by hydrolyzing PPi and providing Pi. In HPP, Phase 1 is normal, but high PPi levels due to deficient TNAP inhibit skeletal matrix mineralization. Elevated phosphorylated OPN also inhibits mineralization in HPP mouse models.
(b) Severe and Mild HPP Cases: Severe HPP: Blocked calcium and Pi entry into the skeleton elevates serum levels, reduces PTH, and causes hypercalciuria. Mild HPP: Elevated Pi levels are due to increased renal phosphate reabsorption. TNAP may directly affect renal phosphate reabsorption, compete with PPi, or involve altered phosphatonin levels.
(c) Vitamin B6 Metabolism and CNS: Only pyridoxal 5'-phosphate (PLP) can act as a cofactor in the CNS. PLP must be dephosphorylated to pyridoxal (PL) by TNAP to cross the blood-brain barrier. Inside cells, PL is rephosphorylated to PLP by pyridoxal kinase, which is essential for GABA synthesis.
1.1. Genetic and Molecular Mechanisms Underlying Hypophosphatasia (HPP)
HPP is a metabolic disorder caused by mutations in the ALPL gene, which encodes the TNSALP. TNSALP is an enzyme crucial for bone mineralization, playing a vital role in the hydrolysis of organic pyrophosphate to inorganic phosphate. Mutations in the ALPL gene can produce an abnormal or insufficient amount of functional TNSALP, resulting in the accumulation of pyrophosphate in the body. This accumulation inhibits bone and tooth mineralization, leading to the clinical manifestations of HPP
| [8] | Gasque, K. C. S.; Foster, B. L.; Kuss, P.; Yadav. M. C.; et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl-/- mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. 2015, 72, 137–147. https://doi.org/10.1016/j.bone.2014.11.017 |
| [9] | Millán, J. L.; Whyte, M. P. Alkaline Phosphatase and Hypophosphatasia. Calcif Tissue Int. 2016, 98, 398-416. https://doi.org/10.1007/s00223-015-0079-1 |
[8, 9]
.
The genetic basis of HPP is highly variable, with over 300 mutations identified in the ALPL gene. These mutations can affect different parts of the gene, leading to a spectrum of disease severity. The most common mutation is a deletion of the entire gene, but other mutations, including missense, nonsense, frame-shift, and splice-site mutations, have also been reported. Each mutation contributes to the heterogeneity observed in HPP phenotypes, ranging from perinatal lethality to adult-onset milder symptoms
| [8] | Gasque, K. C. S.; Foster, B. L.; Kuss, P.; Yadav. M. C.; et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl-/- mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. 2015, 72, 137–147. https://doi.org/10.1016/j.bone.2014.11.017 |
| [9] | Millán, J. L.; Whyte, M. P. Alkaline Phosphatase and Hypophosphatasia. Calcif Tissue Int. 2016, 98, 398-416. https://doi.org/10.1007/s00223-015-0079-1 |
| [10] | Khan, A. A.; Brandi, M. L.; Rush, E. T.; Ali, D. S.; et al. Hypophosphatasia diagnosis: current state of the art and proposed diagnostic criteria for children and adults. Osteoporos Int. 2024, 35, 431–438. https://doi.org/10.1007/s00198-023-06844-1 |
[8-10]
.
1.2. Epidemiology, Including Prevalence and Incidence Rates, with a Focus on Adult-onset HPP
The exact prevalence and incidence rates of HPP are challenging to determine due to the condition's rarity and variations in diagnostic criteria. Globally, HPP is estimated to affect approximately one in 20,000 individuals, although this figure likely underestimates the actual number of cases due to underdiagnosis and misdiagnosis, especially in adults. Adult-onset HPP is less commonly diagnosed than pediatric forms, partly because the symptoms may be less pronounced or mistaken for other conditions.
The incidence rate of HPP varies depending on the population studied and the method of ascertainment. In regions with well-established newborn screening programs, the incidence of severe forms of HPP (such as perinatal lethal HPP) may be lower due to the identification and intervention for milder forms during infancy. However, the incidence of adult-onset HPP remains largely unknown, as it is often not systematically recorded or recognized
| [5] | Seefried, L.; Dahir, K.; Petryk, A.; Högler, W.; Linglart, A.; et al. Burden of Illness in Adults With Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Bone Miner Res. 2020, 35, 2171–2178. https://doi.org/10.1002/jbmr.4130 |
| [6] | Kishnani, P.; Petryk, A.; Hoegler, W.; Linglart, A.; et al. SUN-529 Burden of Illness in Adults with Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Endocr Soc. 2019, 3, SUN-529. https://doi.org/10.1210/js.2019-SUN-529 |
| [10] | Khan, A. A.; Brandi, M. L.; Rush, E. T.; Ali, D. S.; et al. Hypophosphatasia diagnosis: current state of the art and proposed diagnostic criteria for children and adults. Osteoporos Int. 2024, 35, 431–438. https://doi.org/10.1007/s00198-023-06844-1 |
[5, 6, 10]
.
1.3. Clinical Spectrum and Heterogeneity of Disease Presentations in Adult
In adults, HPP can present with a wide range of symptoms, from asymptomatic individuals to those experiencing osteomalacia, dental problems, and occasionally fractures. The clinical spectrum of adult-onset HPP reflects the heterogeneity of the disease, influenced by the type and location of the ALPL gene mutation, as well as environmental factors. Clinical presentation of adults with HPP are provided below:
Asymptomatic: Some adults with HPP may remain undiagnosed throughout their lives, especially if they carry a milder form of the disease.
Osteomalacia: Characterized by bone pain, muscle weakness, and fractures, typically occurring in the weight-bearing bones of the legs.
Dental Problems: Premature loss of baby teeth and delayed eruption of permanent teeth, along with periodontal disease, are common.
Fractures: Increased susceptibility to fractures, particularly stress fractures, due to decreased bone mineral density.
The heterogeneity of HPP presentations in adults underscores the importance of a comprehensive clinical evaluation, including family history, physical examination, and laboratory tests to assess ALP activity and genetic testing to confirm the diagnosis. Early diagnosis and appropriate management are crucial for improving the quality of life and preventing complications in adults with HPP
| [10] | Khan, A. A.; Brandi, M. L.; Rush, E. T.; Ali, D. S.; et al. Hypophosphatasia diagnosis: current state of the art and proposed diagnostic criteria for children and adults. Osteoporos Int. 2024, 35, 431–438. https://doi.org/10.1007/s00198-023-06844-1 |
| [11] | Watanabe, A.; Satoh, S.; Fujita, A.; Naing, B. T.; Orimo, H.; Shimada, T. Perinatal hypophosphatasia caused by uniparental isodisomy. Bone. 2014, 60. 93–97. https://doi.org/10.1016/j.bone.2013.12.009 |
[10, 11]
.
2. Current Diagnostic Criteria and Tools for HPP in Adults
HPP diagnosis in adults is challenging due to the heterogeneous presentation of the disease, which can mimic more common conditions. The current diagnostic criteria for HPP include clinical features, biochemical markers, genetic testing, and imaging studies.
Adults with HPP often present with a history of premature tooth loss, recurrent fractures, or chronic musculoskeletal pain. The adult form of HPP can range from mild to severe, making it essential to recognize subtle signs that may not be immediately associated with metabolic bone disease. Recognizing a combination of symptoms, such as stress fractures in the feet, unexplained pain, and a family history of similar symptoms, can be crucial.
Biochemical markers: Key biochemical markers include low serum alkaline phosphatase (ALP) activity, elevated substrates of ALP such as pyridoxal 5’-phosphate (PLP), and phosphoethanolamine (PEA). Low ALP is a hallmark of HPP, but normal levels can occasionally be observed, especially in milder forms or due to laboratory variability. Elevated PLP, a form of vitamin B6, is a more specific marker for HPP when ALP levels are ambiguous. Routine blood tests, however, might not always include these particular markers, leading to missed diagnoses.
Genetic testing: Genetic testing for mutations in the ALPL gene, which encodes TNSALP, is critical for confirming the diagnosis of HPP. Over 300 mutations have been identified, contributing to the disease's variability. Genetic testing helps distinguish HPP from other conditions with similar clinical and biochemical profiles. It also aids in identifying carriers and informing family members about potential genetic risks.
Imaging studies: Imaging studies such as X-rays, bone scintigraphy, and MRI can reveal characteristic features of HPP, including metaphyseal dysplasia, pseudofractures, and osteopenia. Advanced imaging techniques like dual-energy X-ray absorptiometry (DEXA) scans may show low bone mineral density. Still, these findings are non-specific and can overlap with other conditions like osteoporosis
| [8] | Gasque, K. C. S.; Foster, B. L.; Kuss, P.; Yadav. M. C.; et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl-/- mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. 2015, 72, 137–147. https://doi.org/10.1016/j.bone.2014.11.017 |
| [12] | Baumgartner-Sigl, S.; Haberlandt, E.; Mumm, S.; Scholl-Bürgi, S.; et al. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c. 677T > C, p. M226T; c. 1112C > T, p. T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007, 40, 1655–1661. https://doi.org/10.1016/j.bone.2007.01.020 |
| [13] | Whyte, M. P.; Greenberg, C. R.; Salman, N. J.; Bober, MB.; et al. Enzyme-Replacement Therapy in Life-Threatening Hypophosphatasia. N Engl J Med. 2012, 366, 904–913. https://doi.org/10.1056/NEJMoa1106173 |
| [14] | Khandwala, H. M.; Mumm, S.; Whyte, M. P. Low serum alkaline phosphatase activity and pathologic fracture: Case report and brief review of hypophosphatasia diagnosed in adulthood. Endocr Pract. 2006, 12, 676–681. https://doi.org/10.4158/EP.12.6.676 |
[8, 12-14]
.
Common misdiagnoses and differential diagnose
The clinical overlap of HPP with other disorders often leads to misdiagnosis. Conditions frequently mistaken for HPP include:
Osteoporosis: Patients with HPP may receive bisphosphonates, a standard osteoporosis treatment contraindicated in HPP due to worsening bone mineralization.
Osteomalacia: Like HPP, osteomalacia involves defective bone mineralization but typically presents with elevated ALP levels, helping to distinguish between the two.
Rheumatoid Arthritis and Other Rheumatic Diseases: Chronic joint pain in HPP can be misinterpreted as inflammatory arthritis, leading to inappropriate treatments that do not address the underlying metabolic defect.
Differentiating HPP from these conditions requires a thorough clinical evaluation, including a detailed patient history, biochemical analysis, and targeted genetic testing
| [15] | Bangura, A.; Wright, L.; Shuler, T.; Hypophosphatasia: Current Literature for Pathophysiology, Clinical Manifestations, Diagnosis, and Treatment. Cureus. 2020, 12, e8594. https://doi.org/10.7759/cureus.8594 |
| [16] | Whyte, M. P. Hypophosphatasia-aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016, 12, 233-246. https://doi.org/10.1038/nrendo.2016.14 |
[15, 16]
.
3. Role of Genetic Testing, Biochemical Markers, and Imaging in the Diagnosis Process
Genetic testing provides a definitive diagnosis by identifying ALPL gene mutations. A pathogenic mutation confirms HPP and can help stratify the risk among family members (
Figure 2). Genetic counseling is often recommended to discuss the implications of the findings and potential family planning considerations.
Biochemical markers: Biochemical markers play a vital role in the initial screening and diagnosis of HPP. Low ALP activity and elevated PLP and PEA levels strongly suggest HPP. These markers are essential for distinguishing HPP from other metabolic bone diseases and guiding further genetic testing.
Imaging: Imaging complements biochemical and genetic testing by providing visual evidence of bone abnormalities typical of HPP. While DEXA scans can indicate low bone density, specific radiographic findings like pseudofractures and metaphyseal dysplasia are more indicative of HPP. MRI can be particularly useful in assessing bone quality and detecting subtle fractures not visible on X-rays
| [15] | Bangura, A.; Wright, L.; Shuler, T.; Hypophosphatasia: Current Literature for Pathophysiology, Clinical Manifestations, Diagnosis, and Treatment. Cureus. 2020, 12, e8594. https://doi.org/10.7759/cureus.8594 |
[15]
.
Evaluation of limitations and gaps in current diagnostic practices
Under recognition and misdiagnosis: The rarity of HPP and its clinical overlap with more common conditions lead to underdiagnosis. Many clinicians are not familiar with HPP, resulting in misdiagnosis and inappropriate treatment.
Variability in clinical presentation: HPP's wide range of symptoms and severity complicates diagnosis. Milder forms may go unnoticed or be mistaken for other musculoskeletal disorders.
Biochemical variability: ALP levels can vary, and normal ranges may not account for age, sex, and individual variability. Additionally, routine laboratory tests may not include specific markers like PLP.
Genetic testing accessibility: Genetic testing may not be readily available in all healthcare settings, and the interpretation of results requires specialized knowledge. The cost of genetic testing can also be a barrier.
Lack of awareness: Healthcare providers and patients generally lack awareness about HPP, its symptoms, and the importance of early diagnosis and appropriate treatment.
Gaps need for comprehensive guidelines: There is a need for standardized, comprehensive guidelines for diagnosing and managing adult-onset HPP to ensure consistent and accurate diagnosis across different healthcare settings.
Education and Training: Increased education and training for healthcare providers on recognizing and diagnosing HPP are essential. This includes understanding the clinical presentation, appropriate use of biochemical markers, and the role of genetic testing.
Research and Data Collection: More research is needed to understand the full spectrum of HPP in adults, including epidemiological studies to determine its true prevalence. Better data collection on patient outcomes and treatment efficacy will also inform clinical practice.
In conclusion, diagnosing hypophosphatasia in adults requires a multifaceted approach involving clinical assessment, biochemical testing, genetic analysis, and imaging studies. Addressing the current limitations and gaps in diagnostic practices through education, comprehensive guidelines, and research will improve diagnostic accuracy and patient outcomes, ensuring that individuals with HPP receive the appropriate care
| [1] | Simon, S.; Resch, H.; Klaushofer, K.; Roschger, P.; Zwerina, J.; Kocijan, R. Hypophosphatasia: From Diagnosis to Treatment. Curr Rheumatol Rep. 2018, 20, 69. https://doi.org/10.1007/s11926-018-0778-5 |
| [15] | Bangura, A.; Wright, L.; Shuler, T.; Hypophosphatasia: Current Literature for Pathophysiology, Clinical Manifestations, Diagnosis, and Treatment. Cureus. 2020, 12, e8594. https://doi.org/10.7759/cureus.8594 |
| [18] | Bishop, N. Clinical management of hypophosphatasia. Clin. Cases Miner. Bone Metab. 2015, 12, 170–173. https://doi.org/10.11138/ccmbm/2015.12.2.170 |
[1, 15, 18]
.
4. Current and Emerging Treatment Strategies for HPP
4.1. Traditional Management Approaches and Their Limitations
In adult HPP management, traditional approaches have centered around alleviating symptoms and providing supportive care, given the absence of targeted therapies aimed at the root enzymatic defect until recent developments. The cornerstone of these traditional strategies encompasses symptomatic treatment pain management through analgesics like nonsteroidal anti-inflammatory drugs (NSAIDs) and opioids, albeit with caution regarding the latter's potential for dependency and side effects as shown in
Table 1. Bisphosphonates, once erroneously applied under the misconception of HPP resembling osteoporosis, are now recognized as contraindicated due to their propensity to aggravate bone mineralization issues. Orthopedic interventions, including fracture management via casting and surgical fixation, alongside joint replacement surgeries in severe cases of joint deterioration, represent another facet of care.
However, these interventions are often complicated by the poor bone quality and delayed healing characteristics of HPP patients. Physical therapy and rehabilitation, featuring customized exercise programs designed to enhance mobility and muscle strength, play a crucial role but must be meticulously planned to prevent exacerbation of bone pain or risk of fractures. Despite these efforts, traditional management strategies fail to address the underlying cause of HPP, resulting in limited efficacy in halting disease progression. The misuse of bisphosphonates underscores the imperative for precise diagnosis to steer clear of unsuitable treatments. Moreover, the reliance on analgesics and orthopedic measures frequently fails to achieve comprehensive pain relief and functional improvement, leaving patients with a substantial disease burden
| [2] | Tournis, S.; Yavropoulou, M. P.; Polyzos, S. A.; Doulgeraki, A. Hypophosphatasia. J. Clin. Med. 2021, 10, 5676. https://doi.org/10.3390/jcm10235676 |
| [19] | Kishnani, P. S.; Martos-Moreno, G. Á.; Linglart, A.; Petryk, A.; Messali, A.; et al. Effectiveness of asfotase alfa for treatment of adults with hypophosphatasia: results from a global registry. Orphanet J Rare Dis. 2024, 19, 109. https://doi.org/10.1186/s13023-024-03048-6 |
[2, 19]
.
Table 1. Traditional management approaches for hypophosphatasia (HPP) in adults.
Treatment Approach | Description | Benefits | Limitations |
Pain Management | Use of NSAIDs, opioids, and neuropathic pain agents | Provides symptomatic relief from chronic pain | Risk of dependency (opioids), side effects, and incomplete pain relief |
Orthopedic Interventions | Standard procedures for fractures, joint replacements | Manages acute fractures and severe joint damage | Complicated by poor bone quality, delayed healing |
Physical Therapy | Customized exercise programs and fall prevention strategies | Improves mobility, muscle strength, and overall function | Risk of exacerbating bone pain or inducing fractures |
4.2. Role and Efficacy of Enzyme Replacement Therapy (ERT) with Asfotase Alfa
The advent of ERT with asfotase alfa has revolutionized the treatment landscape for HPP. Asfotase alfa is a recombinant form of human TNSALP designed to address the underlying enzyme deficiency in HPP.
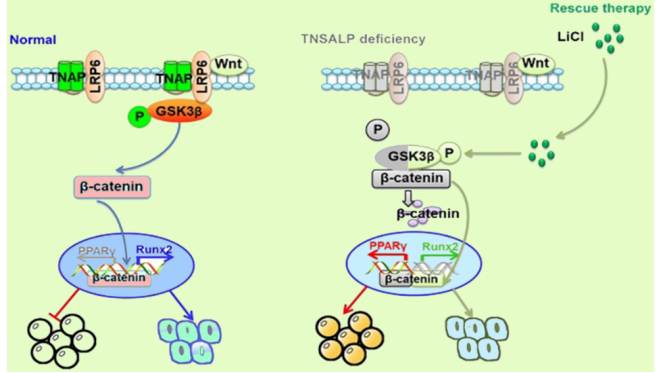
Asfotase alfa works by supplementing deficient alkaline phosphatase, thereby reducing the accumulation of inorganic pyrophosphate and promoting normal bone mineralization. This directly targets the pathophysiological mechanism of HPP, offering a disease-modifying approach rather than merely symptomatic relief
. The possible mechanism of action of Asfotase Alfa in HPP treatment is shown in
Figure 3.
Figure 3. Possible mechanism of action of asfotase alfa in HPP Treatment.
Clinical trials and real-world studies have demonstrated significant benefits of asfotase alfa in adult HPP patients
| [26] | Kishnani, P. S.; Martos-Moreno, G. Á.; Linglart, A.; Petryk, A.; Messali, A.; Fang, S.; Rockman-Greenberg, C.; Ozono, K.; Högler, W.; Seefried, L.; Dahir, K. M. Effectiveness of asfotase alfa for treatment of adults with hypophosphatasia: results from a global registry. Orphanet journal of rare diseases, 2024; 19(1), 109. https://doi.org/10.1186/s13023-024-03048-6 |
[26]
:
Reduction in Fractures: Asfotase alfa has been shown to reduce the frequency of fractures and improve the healing of existing fractures.
Pain Relief: Patients report substantial reductions in bone pain, enhancing their quality of life.
Improved Mobility and Function: Enhanced bone quality and strength improve physical function and mobility.
Overall Skeletal Health: Long-term use of asfotase alfa supports sustained improvements in skeletal health, reducing the risk of complications associated with poor bone mineralization.
Pain Management:
Adjunctive Analgesics: While ERT addresses the underlying cause, additional analgesics may still be needed for residual pain management.
Neuropathic Pain Agents: Medications such as gabapentin or pregabalin may be used for nerve-related pain.
Physical Therapy: Customized Exercise Programs: Ongoing physical therapy tailored to the patient’s capabilities and limitations helps maintain muscle strength, joint function, and overall mobility. Physical therapists can provide training on balance and coordination to reduce the risk of falls and subsequent fractures.
Surgical Interventions: Despite improvements with ERT, surgical management of fractures may still be necessary. Surgeons must be aware of the unique bone properties in HPP to optimize outcomes. For patients with severe joint damage, joint replacement surgeries may be more successful with improved bone quality under ERT.
Asfotase alfa is currently approved for treating perinatal, infantile, and juvenile-onset HPP, where it helps restore enzyme activity and improves bone mineralization. However, its efficacy in late-stage HPP remains a challenge, as the treatment is less effective in patients with advanced bone deformities or severe mineralization defects. In these cases, the therapeutic benefits may be limited, and further research is needed to explore alternative strategies for managing late-stage HPP.
4.3. Assessing the Potential of Emerging Therapies and Ongoing Clinical Trial
4.3.1. Gene Therapy
Mechanism: Gene therapy for HPP aims to correct the underlying genetic defect by delivering functional copies of the ALPL gene to patients' cells. This approach targets the root cause of the disease, potentially providing a long-term or permanent solution. Gene therapy for HPP is in the early stages of development, with preclinical studies showing promise. These studies involve delivering the ALPL gene using viral vectors to correct the enzyme deficiency in animal models. If successful, gene therapy could offer a one-time treatment that restores normal enzyme function, reduces or eliminates the need for ongoing treatments like ERT, and addresses the root cause of HPP.
Challenges: Significant challenges include ensuring safe and effective gene delivery, long-term therapeutic gene expression, and avoiding immune responses to the viral vectors or the introduced gene product
| [13] | Whyte, M. P.; Greenberg, C. R.; Salman, N. J.; Bober, MB.; et al. Enzyme-Replacement Therapy in Life-Threatening Hypophosphatasia. N Engl J Med. 2012, 366, 904–913. https://doi.org/10.1056/NEJMoa1106173 |
| [21] | Buchet, R.; Millán, J. L.; Magne, D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol. 2013, 1053, 27-51. https://doi.org/10.1007/978-1-62703-562-0_3 |
[13, 21]
.
4.3.2. Small Molecule Therapies
Mechanism: Small molecule therapies aim to enhance residual ALP activity or stabilize the mutant enzyme in HPP patients. These molecules could increase the enzyme's effectiveness or prevent its degradation. Research into small molecule therapies is in the preclinical and early clinical trial stages. Various compounds are being investigated for their ability to enhance ALP activity or stabilize the enzyme. Small molecule therapies could provide an oral treatment option that is less invasive and more convenient than ERT. They may also be more affordable, improving accessibility for patients.
Challenges: Identifying effective compounds and ensuring their safety and efficacy in humans are significant hurdles. Additionally, these therapies must be carefully tailored to avoid off-target effects and adverse reactions
| [2] | Tournis, S.; Yavropoulou, M. P.; Polyzos, S. A.; Doulgeraki, A. Hypophosphatasia. J. Clin. Med. 2021, 10, 5676. https://doi.org/10.3390/jcm10235676 |
| [5] | Seefried, L.; Dahir, K.; Petryk, A.; Högler, W.; Linglart, A.; et al. Burden of Illness in Adults With Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Bone Miner Res. 2020, 35, 2171–2178. https://doi.org/10.1002/jbmr.4130 |
| [22] | Hofmann, C.; Seefried, L.; Jakob, F. Asfotase alfa: enzyme replacement for the treatment of bone disease in hypophosphatasia. Drugs Today (Barc). 2016, 52, 271-285. https://doi.org/10.1358/dot.2016.52.5.2482878 |
[2, 5, 22]
.
4.3.3. Combination Therapies
Mechanism: Combining ERT with other therapeutic modalities, such as bone anabolic agents or anti-resorptive drugs, aims to enhance overall treatment efficacy and address residual symptoms that ERT alone may not fully resolve. Several clinical trials are exploring the effectiveness of combination therapies. These studies are investigating the synergistic effects of combining ERT with drugs that promote bone formation or inhibit bone resorption. Combination therapies could provide a more comprehensive approach to managing HPP, improving bone density, reducing fracture risk, and enhancing overall patient outcomes.
ERT has emerged as a pivotal treatment for various genetic disorders characterized by enzyme deficiencies, particularly lysosomal storage diseases and certain metabolic conditions. Since the 1990s, ERT has been successfully applied to treat many rare diseases, most of them being lysosomal storage diseases or metabolic diseases. The production of therapeutic enzymes primarily utilizes mammalian cell cultures, with some derived from plant cells and yeasts, followed by processing to enhance activity, bioavailability, and stability. Challenges in optimizing ERT include improving enzyme interaction with cell membranes, facilitating cellular uptake, reducing immunogenic responses, and overcoming the blood-brain barrier when targeting neuronal cells. Despite these challenges, ERT has demonstrated efficacy and safety, significantly improving patient quality of life and survival rates. Ongoing research focuses on enhancing delivery mechanisms and expanding the range of treatable conditions through ERT
.
HPP is a metabolic disorder resulting from reduced or absent activity of the TNSALP enzyme, due to pathogenic variants in the
ALPL gene. The clinical presentation of HPP varies widely, ranging from severe forms in neonates and infants to milder manifestations in adults. Diagnosing HPP in adults is particularly challenging because symptoms can be mild and non-specific, leading to underdiagnosis or misdiagnosis, especially among clinicians unfamiliar with this rare condition. The absence of formal diagnostic guidelines for adults further complicates consistent diagnosis. To address this, the HPP International Working Group conducted a literature review, identifying six diagnostic findings with a prevalence over 50%, which were considered for major diagnostic criteria. After discussions, they proposed four major and five minor diagnostic criteria for adult HPP. They recommend that a diagnosis in adults should include either two major criteria or one major and two minor criteria
| [24] | Brandi, M. L.; Khan, A. A.; Rush, E. T. et al. The challenge of hypophosphatasia diagnosis in adults: results from the HPP International Working Group Literature Surveillance. Osteoporos Int 2024; 35, 439–449. https://doi.org/10.1007/s00198-023-06859-8 |
[24]
.
HPP is a rare metabolic disorder caused by pathogenic variants in the
ALPL gene, leading to low activity of TNSALP. Diagnosis remains challenging due to the lack of formal guidelines for children, adolescents, and adults. The International HPP Working Group identified key diagnostic criteria, with the highest agreement on pathogenic
ALPL variants, elevated natural substrates, and early loss of primary teeth, organizing these into three major and six minor criteria for diagnosis
| [25] | Rush, E., Brandi, M., Khan, A. et al. Proposed diagnostic criteria for the diagnosis of hypophosphatasia in children and adolescents: results from the HPP International Working Group. Osteoporos Int 2024; 35, 1–10. https://doi.org/10.1007/s00198-023-06843-2 |
[25]
.
Challenges: Determining the optimal combination of therapies, dosages, and treatment regimens is complex. There is also a need to monitor for potential drug interactions and cumulative side effects.
Ongoing clinical trials explore several promising treatment strategies for HPP. Gene therapy trials aim to evaluate the safety, efficacy, and long-term benefits of delivering functional ALPL genes to patients, with successful outcomes potentially leading to regulatory approval and widespread use. Small molecule therapy trials focus on identifying compounds that enhance ALP activity or stabilize the mutant enzyme, with early-phase trials assessing safety and dosage and later-phase trials evaluating the efficacy and long-term benefits. Positive results could result in new, convenient oral medications for HPP. Combination therapy trials investigate the synergistic effects of combining ERT with other drugs, assessing bone density, fracture rates, and quality of life improvements. Success in these trials could establish new standard-of-care protocols incorporating combination therapies for optimal HPP management
.
5. Conclusion
The diagnosis and treatment of HPP in adults present significant challenges, but recent advances offer promising solutions. Diagnostic difficulties arise due to the variability of symptoms and the need for specialized tests, such as genetic analysis and specific biochemical markers. Despite these challenges, ERT with Asfotase alfa has revolutionized treatment, significantly improving patient outcomes by addressing the enzyme deficiency at the root of the disease. However, ERT's high cost, long-term safety concerns, and variability in patient response highlight the need for ongoing research and development of alternative therapies. Emerging treatments, including gene therapy, small molecule therapies, and combination therapies, are currently under investigation in clinical trials. Gene therapy aims to provide a long-term solution by correcting the underlying genetic defect, while small molecule therapies offer the potential for less invasive, more affordable treatment options. Combination therapies seek to enhance the effectiveness of ERT and provide comprehensive management of the disease. Ongoing clinical trials are critical in evaluating these innovative therapies' safety, efficacy, and long-term benefits. Successful trials could lead to new standard-of-care protocols, making advanced treatments more accessible and practical for HPP patients. As research progresses, these advancements hold the potential to significantly improve the diagnosis, management, and overall quality of life for adults living with hypophosphatasia.
Abbreviations
HPP | Hypophosphatasia |
ERT | Enzyme Replacement Therapy |
ALP | Alkaline Phosphatase |
NSAISs | Nonsteroidal Anti- Inflammatory Drugs |
PLP | Pyridoxal 5’- Phosphate |
PEA | Phosphoethanolamine |
TNSALP | Tissue Non- Specific Alkaline Phosphatase |
Author Contributions
Sandeep Bolla is the sole author. The author read and approved the final manuscript.
Funding
This research received no external funding.
Disclaimer
This manuscript reflects the views of the authors and should not be construed to represent views or policies of the affiliated institutions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
| [1] |
Simon, S.; Resch, H.; Klaushofer, K.; Roschger, P.; Zwerina, J.; Kocijan, R. Hypophosphatasia: From Diagnosis to Treatment. Curr Rheumatol Rep. 2018, 20, 69.
https://doi.org/10.1007/s11926-018-0778-5
|
| [2] |
Tournis, S.; Yavropoulou, M. P.; Polyzos, S. A.; Doulgeraki, A. Hypophosphatasia. J. Clin. Med. 2021, 10, 5676.
https://doi.org/10.3390/jcm10235676
|
| [3] |
Martos-Moreno, G. Á.; Calzada, J.; Couce, M. L.; Argente, J. Hypophosphatasia: Clinical manifestations, diagnostic recommendations and therapeutic options. An Pediatría. 2018, 88, 356. e1-356. e11.
https://doi.org/10.1016/j.anpede.2017.06.006
|
| [4] |
Mornet, E.; Taillandier, A.; Domingues, C.; Dufour, A.; Benaloun. E.; et al. Hypophosphatasia: a genetic-based nosology and new insights in genotype-phenotype correlation. Eur J Hum Genet. 2021, 29, 289–299.
https://doi.org/10.1038/s41431-020-00732-6
|
| [5] |
Seefried, L.; Dahir, K.; Petryk, A.; Högler, W.; Linglart, A.; et al. Burden of Illness in Adults With Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Bone Miner Res. 2020, 35, 2171–2178.
https://doi.org/10.1002/jbmr.4130
|
| [6] |
Kishnani, P.; Petryk, A.; Hoegler, W.; Linglart, A.; et al. SUN-529 Burden of Illness in Adults with Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Endocr Soc. 2019, 3, SUN-529.
https://doi.org/10.1210/js.2019-SUN-529
|
| [7] |
González-Cejudo, T.; Villa-Suárez, J. M.; Ferrer-Millán, M.; Andújar-Vera, F.; et al. Mild hypophosphatasia may be twice as prevalent as previously estimated: an effective clinical algorithm to detect undiagnosed cases. Clin Chem Lab Med. 2024, 62, 128–137.
https://doi.org/10.1515/cclm-2023-0427
|
| [8] |
Gasque, K. C. S.; Foster, B. L.; Kuss, P.; Yadav. M. C.; et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl-/- mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. 2015, 72, 137–147.
https://doi.org/10.1016/j.bone.2014.11.017
|
| [9] |
Millán, J. L.; Whyte, M. P. Alkaline Phosphatase and Hypophosphatasia. Calcif Tissue Int. 2016, 98, 398-416.
https://doi.org/10.1007/s00223-015-0079-1
|
| [10] |
Khan, A. A.; Brandi, M. L.; Rush, E. T.; Ali, D. S.; et al. Hypophosphatasia diagnosis: current state of the art and proposed diagnostic criteria for children and adults. Osteoporos Int. 2024, 35, 431–438.
https://doi.org/10.1007/s00198-023-06844-1
|
| [11] |
Watanabe, A.; Satoh, S.; Fujita, A.; Naing, B. T.; Orimo, H.; Shimada, T. Perinatal hypophosphatasia caused by uniparental isodisomy. Bone. 2014, 60. 93–97.
https://doi.org/10.1016/j.bone.2013.12.009
|
| [12] |
Baumgartner-Sigl, S.; Haberlandt, E.; Mumm, S.; Scholl-Bürgi, S.; et al. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c. 677T > C, p. M226T; c. 1112C > T, p. T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007, 40, 1655–1661.
https://doi.org/10.1016/j.bone.2007.01.020
|
| [13] |
Whyte, M. P.; Greenberg, C. R.; Salman, N. J.; Bober, MB.; et al. Enzyme-Replacement Therapy in Life-Threatening Hypophosphatasia. N Engl J Med. 2012, 366, 904–913.
https://doi.org/10.1056/NEJMoa1106173
|
| [14] |
Khandwala, H. M.; Mumm, S.; Whyte, M. P. Low serum alkaline phosphatase activity and pathologic fracture: Case report and brief review of hypophosphatasia diagnosed in adulthood. Endocr Pract. 2006, 12, 676–681.
https://doi.org/10.4158/EP.12.6.676
|
| [15] |
Bangura, A.; Wright, L.; Shuler, T.; Hypophosphatasia: Current Literature for Pathophysiology, Clinical Manifestations, Diagnosis, and Treatment. Cureus. 2020, 12, e8594.
https://doi.org/10.7759/cureus.8594
|
| [16] |
Whyte, M. P. Hypophosphatasia-aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016, 12, 233-246.
https://doi.org/10.1038/nrendo.2016.14
|
| [17] |
Liu, W.; Zhang, L.; Xuan, K.; Hu, C.; et al. Alkaline phosphatase controls lineage switching of mesenchymal stem cells by regulating the lrp6/gsk3β complex in hypophosphatasia. Theranostics. 2018, 8, 5575–5592.
https://doi.org/10.7150/thno.27372
|
| [18] |
Bishop, N. Clinical management of hypophosphatasia. Clin. Cases Miner. Bone Metab. 2015, 12, 170–173.
https://doi.org/10.11138/ccmbm/2015.12.2.170
|
| [19] |
Kishnani, P. S.; Martos-Moreno, G. Á.; Linglart, A.; Petryk, A.; Messali, A.; et al. Effectiveness of asfotase alfa for treatment of adults with hypophosphatasia: results from a global registry. Orphanet J Rare Dis. 2024, 19, 109.
https://doi.org/10.1186/s13023-024-03048-6
|
| [20] |
Scott, L. J. Asfotase Alfa: A Review in Paediatric-Onset Hypophosphatasia. Drugs. 2016, 76, 255-262.
https://doi.org/10.1007/s40265-015-0535-2
|
| [21] |
Buchet, R.; Millán, J. L.; Magne, D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol. 2013, 1053, 27-51.
https://doi.org/10.1007/978-1-62703-562-0_3
|
| [22] |
Hofmann, C.; Seefried, L.; Jakob, F. Asfotase alfa: enzyme replacement for the treatment of bone disease in hypophosphatasia. Drugs Today (Barc). 2016, 52, 271-285.
https://doi.org/10.1358/dot.2016.52.5.2482878
|
| [23] |
Marchetti, M; Faggiano, S; Mozzarelli, A. Enzyme Replacement Therapy for Genetic Disorders Associated with Enzyme Deficiency. Current Medicinal Chemistry. 2021; 29(3): 489–525.
https://doi.org/10.2174/0929867328666210526144654
|
| [24] |
Brandi, M. L.; Khan, A. A.; Rush, E. T. et al. The challenge of hypophosphatasia diagnosis in adults: results from the HPP International Working Group Literature Surveillance. Osteoporos Int 2024; 35, 439–449.
https://doi.org/10.1007/s00198-023-06859-8
|
| [25] |
Rush, E., Brandi, M., Khan, A. et al. Proposed diagnostic criteria for the diagnosis of hypophosphatasia in children and adolescents: results from the HPP International Working Group. Osteoporos Int 2024; 35, 1–10.
https://doi.org/10.1007/s00198-023-06843-2
|
| [26] |
Kishnani, P. S.; Martos-Moreno, G. Á.; Linglart, A.; Petryk, A.; Messali, A.; Fang, S.; Rockman-Greenberg, C.; Ozono, K.; Högler, W.; Seefried, L.; Dahir, K. M. Effectiveness of asfotase alfa for treatment of adults with hypophosphatasia: results from a global registry. Orphanet journal of rare diseases, 2024; 19(1), 109.
https://doi.org/10.1186/s13023-024-03048-6
|
Cite This Article
-
APA Style
Bolla, S. (2025). Challenges and Advances in Diagnosing and Treating Hypophosphatasia in Adults: A Comprehensive Review. International Journal of Genetics and Genomics, 13(1), 1-9. https://doi.org/10.11648/j.ijgg.20251301.11
 Copy
|
Copy
|
 Download
Download
ACS Style
Bolla, S. Challenges and Advances in Diagnosing and Treating Hypophosphatasia in Adults: A Comprehensive Review. Int. J. Genet. Genomics 2025, 13(1), 1-9. doi: 10.11648/j.ijgg.20251301.11
Copy
|
Download
AMA Style
Bolla S. Challenges and Advances in Diagnosing and Treating Hypophosphatasia in Adults: A Comprehensive Review. Int J Genet Genomics. 2025;13(1):1-9. doi: 10.11648/j.ijgg.20251301.11
Copy
|
Download
-
@article{10.11648/j.ijgg.20251301.11,
author = {Sandeep Bolla},
title = {Challenges and Advances in Diagnosing and Treating Hypophosphatasia in Adults: A Comprehensive Review},
journal = {International Journal of Genetics and Genomics},
volume = {13},
number = {1},
pages = {1-9},
doi = {10.11648/j.ijgg.20251301.11},
url = {https://doi.org/10.11648/j.ijgg.20251301.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijgg.20251301.11},
abstract = {Hypophosphatasia (HPP), a rare metabolic disorder caused by mutations in the ALPL gene, leads to deficient alkaline phosphatase activity and presents unique clinical challenges for adult patients, including fractures, chronic pain, and dental issues. This disorder is often underdiagnosed due to its variable clinical presentation and overlap with other conditions, further complicating timely intervention. Early diagnosis is critical for effective management; however, current diagnostic criteria have limitations, often resulting in delays. Insights from the Global HPP Registry reveal significant disease burden and treatment gaps in treated and untreated adults. These insights underscore the importance of multidisciplinary approaches in addressing HPP-related complications, including musculoskeletal and systemic manifestations. Enzyme replacement therapy (ERT) with asfotase alfa has proven effective in reducing disease burden and improving quality of life. Recent clinical evidence suggests that ERT not only alleviates symptoms but may also prevent disease progression when initiated early. Emerging therapies and alternative regimens like teriparatide dosing adjustments are being explored for their potential benefits. In addition, advanced imaging modalities and biomarker studies are improving diagnostic accuracy and monitoring of therapeutic outcomes. This comprehensive review highlights the significant challenges and advances in diagnosing and treating HPP in adults. Ongoing research aims to enhance diagnosis and treatment through genetic testing and personalized medicine, focusing on identifying and addressing knowledge gaps to improve care for adult HPP patients. Collaborative efforts between researchers, clinicians, and patient advocacy groups are crucial for driving innovation and improving access to care. Continued research and innovation are essential, and healthcare professionals must stay informed about the latest advancements in HPP diagnosis and treatment to ensure optimal patient care. By addressing these challenges, the field can move closer to improving the lives of adults living with HPP.},
year = {2025}
}
Copy
|
Download
-
TY - JOUR
T1 - Challenges and Advances in Diagnosing and Treating Hypophosphatasia in Adults: A Comprehensive Review
AU - Sandeep Bolla
Y1 - 2025/01/22
PY - 2025
N1 - https://doi.org/10.11648/j.ijgg.20251301.11
DO - 10.11648/j.ijgg.20251301.11
T2 - International Journal of Genetics and Genomics
JF - International Journal of Genetics and Genomics
JO - International Journal of Genetics and Genomics
SP - 1
EP - 9
PB - Science Publishing Group
SN - 2376-7359
UR - https://doi.org/10.11648/j.ijgg.20251301.11
AB - Hypophosphatasia (HPP), a rare metabolic disorder caused by mutations in the ALPL gene, leads to deficient alkaline phosphatase activity and presents unique clinical challenges for adult patients, including fractures, chronic pain, and dental issues. This disorder is often underdiagnosed due to its variable clinical presentation and overlap with other conditions, further complicating timely intervention. Early diagnosis is critical for effective management; however, current diagnostic criteria have limitations, often resulting in delays. Insights from the Global HPP Registry reveal significant disease burden and treatment gaps in treated and untreated adults. These insights underscore the importance of multidisciplinary approaches in addressing HPP-related complications, including musculoskeletal and systemic manifestations. Enzyme replacement therapy (ERT) with asfotase alfa has proven effective in reducing disease burden and improving quality of life. Recent clinical evidence suggests that ERT not only alleviates symptoms but may also prevent disease progression when initiated early. Emerging therapies and alternative regimens like teriparatide dosing adjustments are being explored for their potential benefits. In addition, advanced imaging modalities and biomarker studies are improving diagnostic accuracy and monitoring of therapeutic outcomes. This comprehensive review highlights the significant challenges and advances in diagnosing and treating HPP in adults. Ongoing research aims to enhance diagnosis and treatment through genetic testing and personalized medicine, focusing on identifying and addressing knowledge gaps to improve care for adult HPP patients. Collaborative efforts between researchers, clinicians, and patient advocacy groups are crucial for driving innovation and improving access to care. Continued research and innovation are essential, and healthcare professionals must stay informed about the latest advancements in HPP diagnosis and treatment to ensure optimal patient care. By addressing these challenges, the field can move closer to improving the lives of adults living with HPP.
VL - 13
IS - 1
ER -
Copy
|
Download