Zeugodacus cucurbitae or melon fly formerly called Bactrocera cucurbitae is an agricultural pest of Asian origin. Well known as a pest of fleshy fruits and vegetables damaging 81 host plants, the melon fly has been the subject of several studies due to its introduction and dissemination worldwide. Up to now, no study on the global structuring of Zeugodacus cucurbitae has been done. Therefore, knowledge of its genetic structuring would allow better management of the insect. It is in this context that the present study on the genetic characterization of populations of Z. cucurbitae watermelon pest insect fits. Our data was collected from the Genbank database. Phylogeographic analyses were made using mitochondrial cytochrome oxidase I (COI) DNA as a genetic marker. After analysis, the study demonstrated two distinct groups: a group composed of the population of Reunion and another group composed of populations from Africa, Asia, Oceania, and Hawaii. This is the result of a genetic isolation demonstrated by the Mantel Test for which the significant p-value confirms the correlation between genetic distances and geographical distances. However, there is a genetic differentiation between individuals in the Reunion population. For any fight against this insect, it would be interesting to take into account the existence of these two genetic groups.
| Published in | International Journal of Genetics and Genomics (Volume 13, Issue 4) |
| DOI | 10.11648/j.ijgg.20251304.12 |
| Page(s) | 83-94 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Zeugodacus Cucurbitae, Characterisation, COI, Phylogeography, Mitochondrial DNA
E | N | M | I | S=S2V | P | P2V | P3V | P4V | Eta |
|---|---|---|---|---|---|---|---|---|---|
182 | 592 | 247 | 312 | 12 | 300 | 284 | 13 | 3 | 331 |
Bases | A | T | C | G |
|---|---|---|---|---|
A | - | 5.79 | 2.72 | 12.06 |
T | 4.23 | - | 11.38 | 2.29 |
C | 4.23 | 24.20 | - | 2.29 |
G | 22.30 | 5.79 | 2.72 | - |
Total population | Senegal | Tanzania | Thailand | Bangladesh | Sri Lanka | Indonesia | |
|---|---|---|---|---|---|---|---|
hd | 0.390+/-0.002 | 0.2+/- 0.238 | 0.378+/-0.032 | 0.378+/-0.032 | 0.778+/-0.0188 | 0.778+/-0.02 | 0.286+/-0.038 |
Pi | 0.056+/- 0.102 | 0.000+/-0.001 | 0.001+/-0.001 | 0.001+/-0.001 | 0.0017+/-0.003 | 0.003+/-0.004 | 0.0005+/-0.001 |
China | Vietnam | Philippines | Taiwan | Laos | Malaysia | Nepal | |
hd | 0.378+/- 0.033 | 0.778+/-0.019 | 0.200+/-0.024 | 0.378+/-0.033 | 0.200+/-0.024 | 0.222+/-0.028 | 0.833+/-0.016 |
Pi | 0.004+/-0.006 | 0.002+/-0.0036 | 0.000+/-0.0006 | 0.000+/-0.0012 | 0.000+/-0.0006 | 0.000+/-0.0006 | 0.003+/-0.0044 |
Japan | Mariana Islands | Salomon Island | Guam | Hawaii | Reunion | Cambodia | |
hd | 0.400+/-0.0563 | 0.417+/-0.036 | 0.0 | 0.00 | 0.200+/-0.023 | 0.378+/-0.0328 | 0.286+/-0.0385 |
Pi | 0.0006+/-0.0008 | 0.000+/-0.001 | 0.0 | 0.00 | 0.00034+/-0.00060 | 0.00312+/-0.00503 | 0.00049+/-0.00069 |
Nepal | P-value | Reunion | P-value | |
|---|---|---|---|---|
Senegal | 0.012 | 0.013 | 0.997 | 0,00 |
Tanzania | 0.009 | 0.046 | 0.997 | 0.00 |
Thailand | 0.997 | 0.00 | ||
Bangladesh | 0.994 | 0.00 | ||
Sri Lanka | 0.996 | 0.00 | ||
Indonesia | 0.997 | 0.00 | ||
China | 0.997 | 0.00 | ||
Vietnam | 0.996 | 0.00 | ||
Philippines | 0.997 | 0.00 | ||
Taiwan | 0.994 | 0.00 | ||
Laos | 0.09 | 0.039 | 0.996 | 0.00 |
Malaysia | 0.996 | 0.00 | ||
Nepal | 0.996 | 0.00 | ||
Cambodia | 0.996 | 0.00 | ||
Japan | 0.996 | 0.00 | ||
Mariana Islands | 0.997 | 0.00 | ||
Salomon Island | 0.996 | 0.00 | ||
Guam | 0.996 | 0.00 | ||
Hawaii | 0,009 | 0,041 | 0.997 | 0.00 |
Distances | Intragroup distances | Intergroup distances | SD | |
|---|---|---|---|---|
Groups | G2 | 0.001 | 0.213 | 0.028 |
0.017 | ||||
G1 | 0.338 | 0.000 |
Distances | Intragroup distances | Intergroup distances | SD | |
|---|---|---|---|---|
Groups | Reunion | 0.003 | 0.213 | 0.001 |
0.017 | ||||
Pays | 0,001 | 0,000 |
Groups | Distances | SD |
|---|---|---|
Africa | 0.000 | 0.000 |
Asia | 0.001 | 0.000 |
Oceania | 0.000 | 0.000 |
Europe | 0.003 | 0.001 |
US | 0.000 | 0.000 |
Groups | Genetic distances | SD |
|---|---|---|
Africa-Asia | 0.001 | 0.000 |
Africa-Oceania | 0.000 | 0.000 |
Asia-Oceania | 0.001 | 0.000 |
Africa- United States | 0.000 | 0.000 |
Asia- United States | 0.001 | 0.000 |
Oceania- United States | 0.000 | 0.000 |
Africa-Europe | 0.954 | 0.080 |
Asia-Europe | 0.955 | 0.080 |
Oceania-Europe | 0.954 | 0.080 |
United States -Europe | 0.954 | 0.080 |
r (AB) | p-value | alpha |
|---|---|---|
0.185 | 0.012 | 0.05 |
Source of Variation | d.f | Sum of squares | Variance component | Variance percentage |
|---|---|---|---|---|
Between groups | 1 | 3107.501 | 164.39391Va | 99.81 |
Between populations inside groups | 18 | 5.006 | -0.00345Vb | -0.00 |
Inside populations | 162 | 50.103 | 0.30928Vc | 0.19 |
Total | 181 | 3162.610 | 164.69975 | |
Indices of fixation | Fst entre groupes = 0.99812 p value: 0.00000+-0.00000 | |||
Populations | D of Tajima | P value |
|---|---|---|
Bangladesh | -1.66706 | 0.03350 |
China | -1.72953 | 0.02990 |
Vietnam | -1.66706 | 0.02830 |
Nepal | -1.76663 | 0.01700 |
Reunion | -1.63600 | 0.04160 |
Populations | D of Tajima | P value |
|---|---|---|
Bangladesh | 2.84720 | 0.00160 |
Sri Lanka | -2.01642 | 0.04930 |
Vietnam | -1.34464 | 0.04530 |
Taiwan | -1.16394 | 0.03670 |
Nepal | -2.87184 | 0.00820 |
Re | Reunion |
SL | Sri Lanka |
Vt | Vietnam |
Np | Nepal |
Ch | China |
Tz | Tanzania |
Th | Thailand |
Tw | Taiwan |
Bd | Bangladesh |
Ha | Hawaii |
Im | Mariana Islands |
Bdo | Bactrocera dorsalis |
| [1] | Hdider, C., Tlili, I., & Ilahy, R. (2020). Watermelon. In Nutritional Composition and Antioxidant Properties of Fruits and Vegetables. Elsevier. p. 515-531. |
| [2] | Badji, H., & Coly, E. V. (2010). Gestion des Attaques de la Mouche des Fruits sur les Cultures de Cucurbitacées au Moyen de Pesticides Naturels au Sénégal. International Symposium on Urban and Peri-Urban Horticulture in the Century of Cities: Lessons, Challenges, Opportunitites 1021, 421-426. |
| [3] | Kunprom, C., & Pramual, P. (2017). Genetic structure and demographic history of the melon fly Zeugodacus cucurbitae (Coquillet) (Diptera: Tephritidae) in Thailand. Agr. For. Entomol, 20, 180-190. |
| [4] | White, I. M., De Meyer, M. & Stonehouse, J. M. (2000) A review of native and introduced fruit flies (Diptera, Tephritidae) in the Indian Ocean islands of Mauritius, Réunion, and Seychelles. Proceedings, Indian Ocean Commission, Regional Fruit Fly Symposium (ed. by N. S. Price and S. I. Seewooruthun). Indian Ocean Commission/European Union, Flic en Flac, Mauritius. pp. 15-21. |
| [5] | Dhillon, M. K., Singh, R., Naresh, J. S., & Sharma, H. C. (2005). The melon fruit fly, Bactrocera cucurbitae: A review of its biology and management. Journal of Insect Science, 5(1), 40-56. |
| [6] | Bezzi, M. (1913). Indian Trypaneids [Trypetids](Fruit-Flies) in the Collection of the Indian Museum, Calcutta. Memoirs of The Indian Museum, 3, 53-168. |
| [7] | Nishida T, Bess HA (1957). Studies on the ecology and control of the melon fly Dacus (Strumeta) cucurbitae Coquillet (Diptera: Tephritidae). Hawaii Agricultural Experiment Station, University of Hawaii. Tech Bull 84: 12-29. |
| [8] | Virgilio, M., Delatte, H., Backeljau, T., & De Meyer, M. (2010). Macrogeographic population structuring in the cosmopolitan agricultural pest Bactrocera cucurbitae (Diptera: Tephritidae). Molecular Ecology, 19(13), 2713-2724. |
| [9] | Jacquard, C., Virgilio, M., David, P., Quilici, S., De Meyer, M., & Delatte, H. (2013). Population structure of the melon fly, Bactrocera cucurbitae, in Reunion Island. Biological Invasions, 15(4), 759-773. |
| [10] | Delatte, H., De Meyer, M., & Virgilio, M. (2019). Genetic structure and range expansion of Zeugodacus Cucurbitae (Diptera: Tephritidae) in Africa. Bulletin of Entomological Research, 109(6), 713-722. |
| [11] | Hu Jian, H. J., Zhang, J. L., Nardi, F., & Zhang, R. J. (2008). Population genetic structure of the melon fly, Bactrocera cucurbitae (Diptera: Tephritidae), from China and Southeast Asia. 134, 319-324. |
| [12] | Prabhakar, C. S., Mehta, P. K., Sood, P., Singh, S. K., Sharma, P., & Sharma, P. N. (2012). Population genetic structure of the melon fly, Bactrocera cucurbitae (Coquillett) (Diptera: Tephritidae) based on mitochondrial cytochrome oxidase (COI) gene sequences. Genetica, 140(1-3), 83-91. |
| [13] | Wu, Y., McPheron, B. A., Wu, J., & Li, Z. (2012). Genetic relationship of the melon fly, Bactrocera cucurbitae (Diptera: Tephritidae) inferred from mitochondrial DNA. Insect Science, 19(2), 195-204. |
| [14] | Warot, S. F. (2018). Caractérisation moléculaire et isolements reproducteurs chez des auxiliaires de lutte biologique. Mémoire. Université Paris sciences et lettres, Paris. 94p. HAL Id: hal-03236272 |
| [15] | Avise, J. C., Arnold, J., Ball, R. M., Bermingham, E., Lamb, T., Neigel, J. E., Reeb, C. A., & Saunders, N. C. (1987). Intraspecific phylogeography: The Mitochondrial DNA Bridge Between Population Genetics and Systematics. Annual Review of Ecology and Systematics, 18(1), 489-522. |
| [16] | Hebert, P. D. N., Cywinska, A., Ball, S. L., & deWaard, J. R. (2003). Biological identifications through DNA barcodes. Proceedings of the Royal Society of London. Series B: Biological Sciences, 270(1512), 313-321. |
| [17] | Ndiaye, M. R. (2018). Evolution phylogéographique et structure génétique des populations de Sitophilus zeamais (Coleoptera, Curculionidae), ravageur du maïs stocke en Afrique de l’Ouest [Phdthesis, Université Cheikh Anta DIOP de Dakar, Sénégal ; Faculté des Sciences et Techniques; Département de Biologie Animale]. 178p. |
| [18] | Sabir, J. S. M., Rabah, S., Yacoub, H., Hajrah, N. H., Atef, A., Al-Matary, M., Edris, S., Alharbi, M. G., Ganash, M., Mahyoub, J., Al-Hindi, R. R., Al-Ghamdi, K. M., Hall, N., Bahieldin, A., Kamli, M. R., & Rather, I. A. (2019). Molecular evolution of cytochrome C oxidase-I protein of insects living in Saudi Arabia. PLoS ONE, 14(11), 1-18. |
| [19] |
NDONG, A., THIAW, C., Diallo, B., SARR, M., DIOME, T., KANE, M., & SEMBENE, M. (2015). Barcoding: Comparison of Variation Degree of COI and Cytochrome b Mitochondrial Markers in Two Species Primary Maize Pests (Sitophilus zeamais and Sitophilus oryzae). International Journal of Sciences: Basic and Applied Research, 20(1), 373-393.
https://www.gssrr.org/index.php/JournalOfBasicAndApplied/article/view/3420 |
| [20] | Avise, J. C. (2000). Phylogeography: The history and formation of species. Harvard university press. Cambridge, MA. 441p. |
| [21] | Saccone, C., De Giorgi, C., Gissi, C., Pesole, G., & Reyes, A. (1999). Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene, 238(1), 195-209. |
| [22] | Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium series, 41(41), p 95-98. |
| [23] | Librado, P., & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25(11), 1451-1452. |
| [24] | Tamura, K., Stecher, G., Peterson, D., Filipski, A., & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular biology and evolution, 30(12), 2725-2729. |
| [25] | Nei, M. (1973). Analysis of Gene Diversity in Subdivided Populations. Proceedings of the National Academy of Sciences, 70(12), 3321-3323. |
| [26] | Nei, M. (1977). F ‐statistics and analysis of gene diversity in subdivided populations. Annals of Human Genetics, 41(2), 225-233. |
| [27] | Bandelt, H. J., Forster, P., Röhl, A., (1999). Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1), 37-48, |
| [28] | Excoffier, L., & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564-567. |
| [29] | Huelsenbeck, J. P., & Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics, 17(8), 754-755. |
| [30] | Rambaut, A., & Drummond, A. J. (2012). Figtree, a graphical viewer of phylogenetic trees. V. 1.4. 0. Available from |
| [31] | Gasparich, G. E., Silva, J. G., Han, H.-Y., Mcpheron, B. A., Steck, G. J., & Sheppard, W. S. (1997). Population genetic structure of Mediterranean fruit fly (Diptera: Tephritidae) and implications for worldwide colonization patterns. Annals of the Entomological Society of America, 90(6), 790-797. |
| [32] | Denno, R. F., McClure, M. S., & Ott, J. R. (1995). Interspecific interactions in phytophagous insects: Competition reexamined and resurrected. Annual review of entomology, 40(1), 297-331. |
| [33] | Jacquard, C., Virgilio, M., Quilici, S., De Meyer, M., & Delatte, H. (2012). Spatial scales of genetic structuring in Bactrocera cucurbitae (Diptera, Tephritidae): Population structure on la Réunion. |
APA Style
Mendy, M. I., Diome, T., Faye, M., Sembène, M. (2025). Genetic Characterization of Populations of Zeugodacus Cucurbitae (Coquillet, 1899), a Watermelon Pest. International Journal of Genetics and Genomics, 13(4), 83-94. https://doi.org/10.11648/j.ijgg.20251304.12
ACS Style
Mendy, M. I.; Diome, T.; Faye, M.; Sembène, M. Genetic Characterization of Populations of Zeugodacus Cucurbitae (Coquillet, 1899), a Watermelon Pest. Int. J. Genet. Genomics 2025, 13(4), 83-94. doi: 10.11648/j.ijgg.20251304.12
@article{10.11648/j.ijgg.20251304.12,
author = {Madeleine Ivonne Mendy and Toffène Diome and Mamecor Faye and Mbacké Sembène},
title = {Genetic Characterization of Populations of Zeugodacus Cucurbitae (Coquillet, 1899), a Watermelon Pest
},
journal = {International Journal of Genetics and Genomics},
volume = {13},
number = {4},
pages = {83-94},
doi = {10.11648/j.ijgg.20251304.12},
url = {https://doi.org/10.11648/j.ijgg.20251304.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijgg.20251304.12},
abstract = {Zeugodacus cucurbitae or melon fly formerly called Bactrocera cucurbitae is an agricultural pest of Asian origin. Well known as a pest of fleshy fruits and vegetables damaging 81 host plants, the melon fly has been the subject of several studies due to its introduction and dissemination worldwide. Up to now, no study on the global structuring of Zeugodacus cucurbitae has been done. Therefore, knowledge of its genetic structuring would allow better management of the insect. It is in this context that the present study on the genetic characterization of populations of Z. cucurbitae watermelon pest insect fits. Our data was collected from the Genbank database. Phylogeographic analyses were made using mitochondrial cytochrome oxidase I (COI) DNA as a genetic marker. After analysis, the study demonstrated two distinct groups: a group composed of the population of Reunion and another group composed of populations from Africa, Asia, Oceania, and Hawaii. This is the result of a genetic isolation demonstrated by the Mantel Test for which the significant p-value confirms the correlation between genetic distances and geographical distances. However, there is a genetic differentiation between individuals in the Reunion population. For any fight against this insect, it would be interesting to take into account the existence of these two genetic groups.
},
year = {2025}
}
TY - JOUR T1 - Genetic Characterization of Populations of Zeugodacus Cucurbitae (Coquillet, 1899), a Watermelon Pest AU - Madeleine Ivonne Mendy AU - Toffène Diome AU - Mamecor Faye AU - Mbacké Sembène Y1 - 2025/10/14 PY - 2025 N1 - https://doi.org/10.11648/j.ijgg.20251304.12 DO - 10.11648/j.ijgg.20251304.12 T2 - International Journal of Genetics and Genomics JF - International Journal of Genetics and Genomics JO - International Journal of Genetics and Genomics SP - 83 EP - 94 PB - Science Publishing Group SN - 2376-7359 UR - https://doi.org/10.11648/j.ijgg.20251304.12 AB - Zeugodacus cucurbitae or melon fly formerly called Bactrocera cucurbitae is an agricultural pest of Asian origin. Well known as a pest of fleshy fruits and vegetables damaging 81 host plants, the melon fly has been the subject of several studies due to its introduction and dissemination worldwide. Up to now, no study on the global structuring of Zeugodacus cucurbitae has been done. Therefore, knowledge of its genetic structuring would allow better management of the insect. It is in this context that the present study on the genetic characterization of populations of Z. cucurbitae watermelon pest insect fits. Our data was collected from the Genbank database. Phylogeographic analyses were made using mitochondrial cytochrome oxidase I (COI) DNA as a genetic marker. After analysis, the study demonstrated two distinct groups: a group composed of the population of Reunion and another group composed of populations from Africa, Asia, Oceania, and Hawaii. This is the result of a genetic isolation demonstrated by the Mantel Test for which the significant p-value confirms the correlation between genetic distances and geographical distances. However, there is a genetic differentiation between individuals in the Reunion population. For any fight against this insect, it would be interesting to take into account the existence of these two genetic groups. VL - 13 IS - 4 ER -
Department of Animal Biology, Cheikh Anta Diop University, Dakar, Senegal
Department of Animal Biology, Cheikh Anta Diop University, Dakar, Senegal
Department of Animal Biology, Cheikh Anta Diop University, Dakar, Senegal
Department of Animal Biology, Cheikh Anta Diop University, Dakar, Senegal
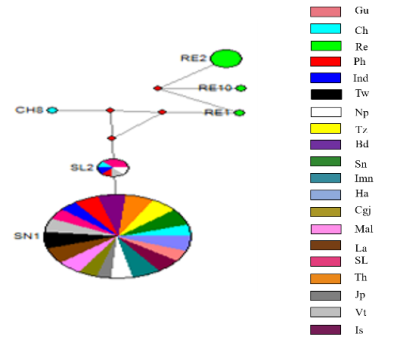
Figure 1. Haplotype network of Zeugodacus cucurbitae. The size of circles is proportional to the number of individuals in the haplotype or the number of haplotypes in the haplogroup.
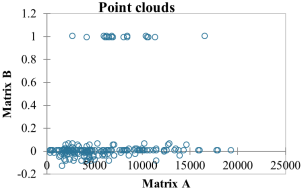
Figure 2. Correlation between geographical distances and Fst of Z. cucurbitae.
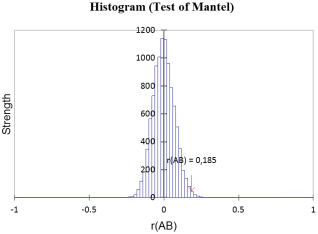
Figure 3. Mantel Pearson-type histogram of Z. cucurbitae.
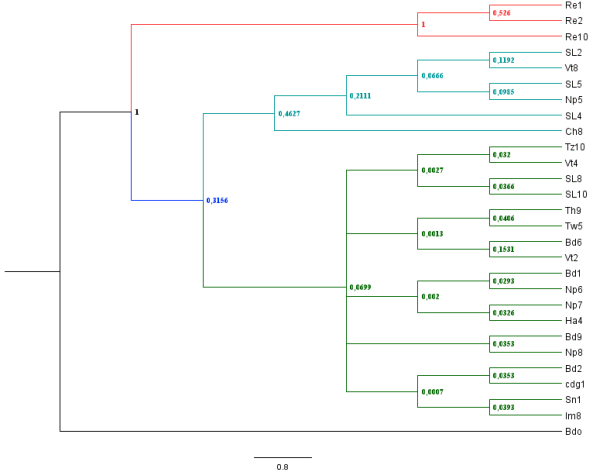
Figure 4. Phylogenetic tree of haplotypes by the Bayesian inference method.
Information

